9: Phosphate transfer reactions
Contents
9: Phosphate transfer reactions#

Mono Lake, California
(photo credit https://www.flickr.com/photos/slolane/)
Introduction#
This chapter is about the chemistry of phosphates, a ubiquitous functional group in biomolecules that is based on phosphoric acid:
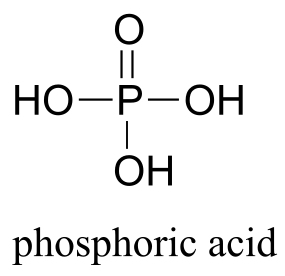
fig 1d
In late 2010, people around the world found themselves getting a crash course in phosphate chemistry as they watched the evening news. Those who paid close attention to the developing story also got an interesting glimpse into the world of scientific research and debate.
It all started when the American National Aeronautics and Space Administration (NASA) released the following statement to the news media:
“NASA will hold a news conference at 2 p.m. EST on Thursday, Dec. 2, to
discuss an astrobiology finding that will impact the search for evidence of
extraterrestrial life.”
The wording of the statement attracted widespread media attention, and had some people holding their breath in anticipation that NASA would be introducing a newly discovered alien life form to the world. When December 2nd came, however, those hoping to meet ET were disappointed – the life form being introduced was a bacterium, and it was from our own planet. To biologists and chemists, though, the announcement was nothing less than astounding.
The NASA scientists worked hard to emphasize the significance of their discovery during the news conference. Dr. Felicia Wolfe-Simon, a young postdoctoral researcher who had spearheaded the project, stated that they had “cracked open the door to what’s possible for life elsewhere in the universe - and that’s profound”. A senior NASA scientist claimed that their results would “fundamentally change how we define life”, and, in attempting to convey the importance of the discovery to a reporter from the newspaper USA Today, referred to an episode from the original Star Trek television series in which the crew of the Starship Enterprise encounters a race of beings whose biochemistry is based on silica rather than carbon.
The new strain of bacteria, dubbed ‘GFAJ-1’, had been isolated from the arsenic-rich mud surrounding salty, alkaline Mono Lake in central California. What made the strain so unique, according to the NASA team, was that it had evolved the ability to substitute arsenate for phosphate in its DNA. Students of biology and chemistry know that phosphorus is one of the six elements that are absolutely required for life as we know it, and that DNA is a polymer linked by phosphate groups. Arsenic, which is directly below phosphorus on the periodic table, is able to assume a bonding arrangement like that of phosphate, so it might seem reasonable to wonder whether arsenate could replace phosphate in DNA and other biological molecules. Actually finding a living thing with arsenate-linked DNA would indeed be a momentous achievement in biology, as this would represent a whole new chemistry for the most fundamental molecule of life, and would change our understanding of the chemical requirements for life to exist on earth - and potentially other planets.
In 1987, Professor F.H. Westheimer of Harvard University published what would become a widely read commentary in Science Magazine entitled “Why Nature Chose Phosphates”. In it, he discussed the chemical properties that make the phosphate group so ideal for the many roles that it plays in biochemistry, chief among them the role of a linker group for DNA polymers. One of the critical characteristics of phosphate that Westheimer pointed out was that the bonds linking phosphate to organic molecules are stable in water. Clearly, if you are selecting a functional group to link your DNA, you don’t want to choose one that will rapidly break apart in water. Among the functional groups that Westheimer compared to phosphate in terms of its suitability as a potential DNA linker was arsenate –but he very quickly dismissed the idea of arsenate-linked DNA because it would be far too unstable in water.
Given this background, it is not hard to imagine that many scientists were puzzled, to say the least, by the NASA results. While the popular media took the announcement at face value and excitedly reported the results as a monumental discovery – NASA is, after all, a highly respected scientific body and the study was being published in Science Magazine, one of the most prestigious scientific journals in the world – many scientists quickly voiced their skepticism, mainly in the relatively new and unconstrained venue of the blogosphere. Microbiologist Rosie Redfield of the University of British Columbia, writing in her blog devoted to ‘open science’, wrote a detailed and highly critical analysis of the study. She pointed out, among other things, that the experimenters had failed to perform the critical purification and mass spectrometry analyses needed to demonstrate that arsenate was indeed being incorporated into the DNA backbone, and that the broth in which the bacteria were being grown actually contained enough phosphate for them to live and replicate using normal phosphate-linked DNA. Science journalist Carl Zimmer, in a column in the online magazine Slate, contacted twelve experts to get their opinions, and they were overwhelmingly negative. One of the experts said bluntly, “This paper should not have been published”. Basically, the NASA researchers were making an astounding claim that, if true, would refute decades of established knowledge about the chemistry of DNA – but the evidence they presented was far from convincing. Carl Sagan’s widely quoted dictum - “extraordinary claims require extraordinary evidence” - seemed to apply remarkably well to the situation.
What followed was a very public, very lively, and not always completely collegial debate among scientists about the proper way to discuss science: the NASA researchers appeared to dismiss the criticism amassed against their study because it came from blogs, websites, and Twitter feeds. The proper venue for such discussion, they claimed, was in the peer-reviewed literature. Critics countered that their refusal to respond to anything outside of the traditional peer-review system was disingenuous, because they had made full use of the publicity-generating power of the internet and mainstream media in the first place when they announced their results with such fanfare.
The traditional venue for debate, while quite a bit slower than the blogosphere, did eventually come through. When the full paper was published in Science a few months later, it was accompanied by eight ‘technical comments’ from other researchers pointing out deficiencies in the study, an ‘editors note’, and a broader news article about the controversy. In July of 2012, a paper was published in Science under the title “GFAJ-1 Is an Arsenate-Resistant, Phosphate-Dependent Organism”. The paper reported definitive evidence that DNA from GFAJ-1, under the conditions described in the NASA paper, did not have arsenate incorporated into its structure. Just like professor Westheimer discussed in the 1980s, it appears that nature really did choose phosphate – and only phosphate – after all … at least on this planet.
Background reading and viewing:
Youtube video of the NASA press conference:
http://www.youtube.com/watch?v=WVuhBt03z8g.
Wolfe-Simon, F. et al. Science Express, Dec 2, 2010. The first preview article on the proposed ‘arsenic bacteria’.
Wolfe-Simon, F. et al., Science 2011, 332, 1163. The full research paper in Science Magazine.
Westheimer, F.H. Science 1987, 235, 1173. The article by Westheimer titled ‘Why Nature Chose Phosphates’.
Zimmer, Carl, Slate, Dec 7, 2010: Blog post by Carl Zimmer titled ‘This Paper Should Not Have Been Published’.
http://www.slate.com/articles/health_and_science/science/2010/12/this_paper_should_not_have_been_published.html
Redfield, R. Blog post Dec 4, 2010:
http://rrresearch.fieldofscience.com/2010/12/arsenic-associated-bacteria-nasas.html
Science 2012, 337, 467. The paper in Science Magazine refuting the validity of the arsenic bacteria claim.
9.1: Overview of phosphate groups#
Phosphate is everywhere in biochemistry. As we were reminded in the introduction to this chapter, our DNA is linked by phosphate:
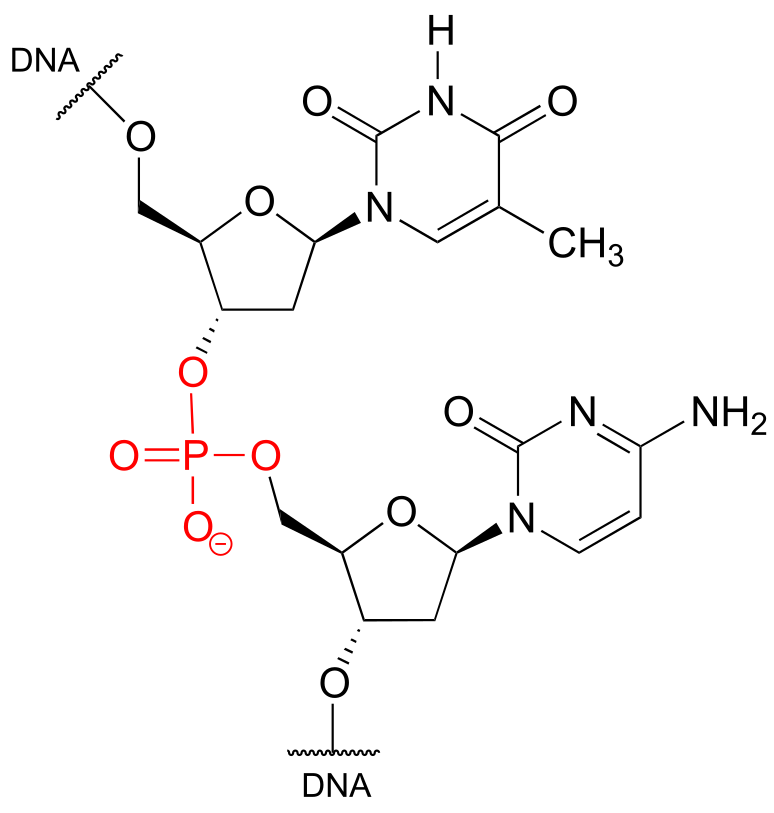
fig 1a
The function of many proteins is regulated - switched on and off - by enzymes which attach or remove a phosphate group from the side chains of serine, threonine, or tyrosine residues.

fig 1b
Countless diseases are caused by defects in phosphate transferring enzymes. As just one example, achondroplasia, a common cause of dwarfism, is caused by a defect in an enzyme whose function is to transfer a phosphate to a tyrosine residue in a growth-related signaling protein.
Finally, phosphates are excellent leaving groups in biological organic reactions, as we will see many times throughout the remainder of this book.
Clearly, an understanding of phosphate chemistry is central to the study of biological organic chemistry. We’ll begin with an overview of terms used when talking about phosphates.
9.1A: Terms and abbreviations#
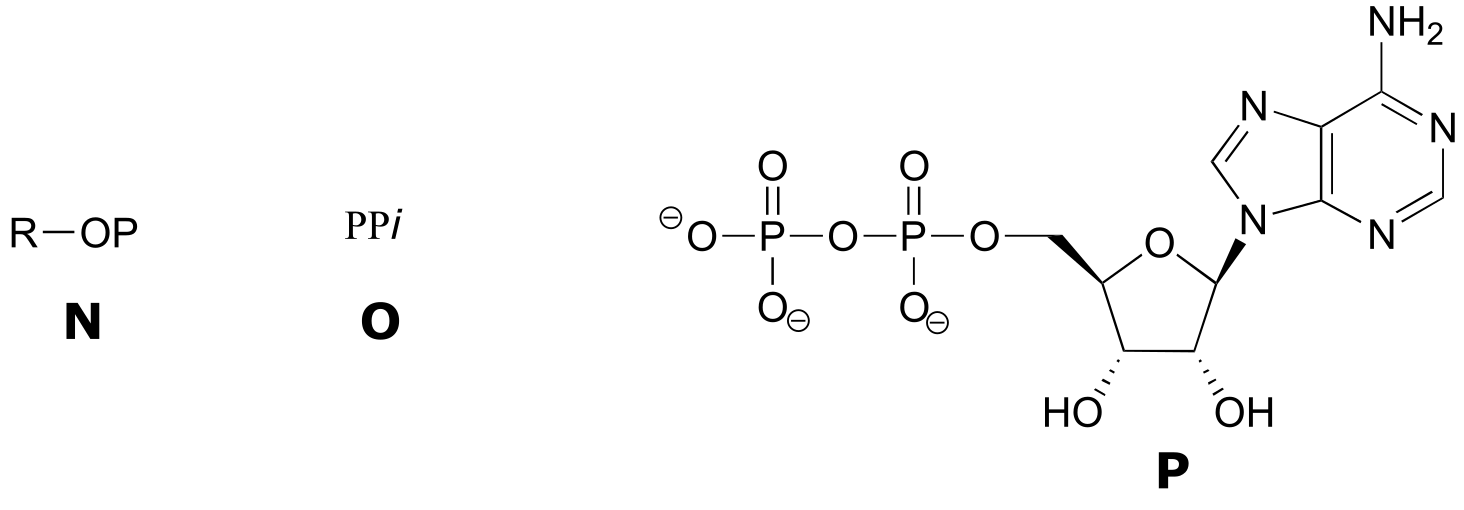
The fully deprotonated conjugate base of phosphoric acid is called a phosphate ion, or inorganic phosphate (often abbreviated ‘P*i’). When two phosphate groups are linked to each other, the linkage itself is referred to as a ‘phosphate anhydride’, and the compound is called ‘inorganic pyrophosphate’ (often abbreviated PPi*).
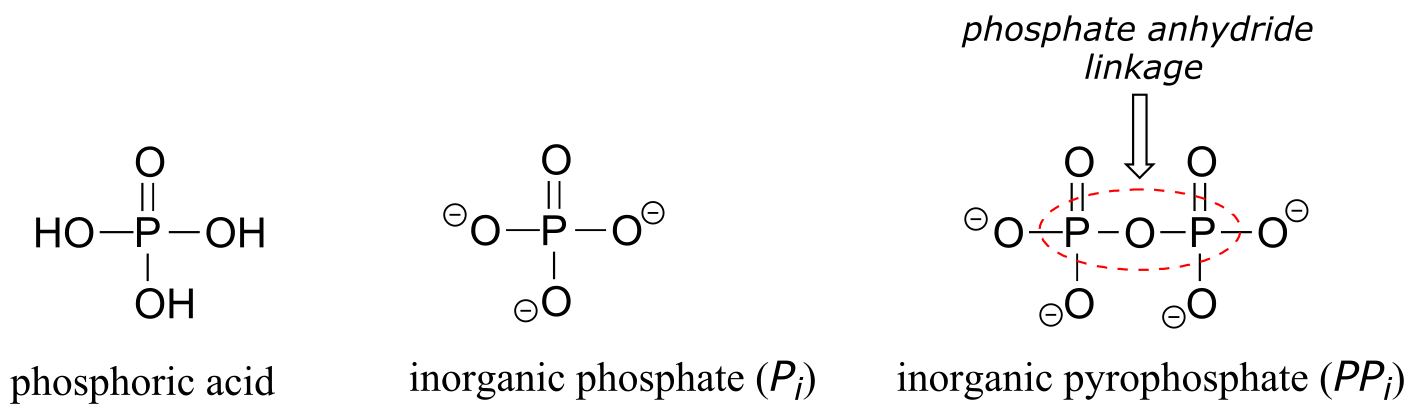
fig 1
The chemical linkage between phosphate and a carbon atom is a phosphate ester. Adenosine monophosphate (AMP) has a single phosphate ester linkage.
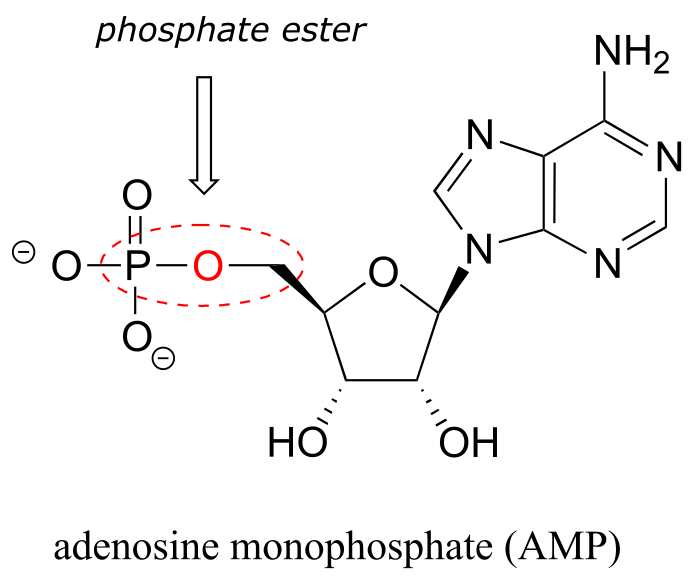
fig 2
Adenosine triphosphate has one phosphate ester linkage and two phosphate anhydride linkages.
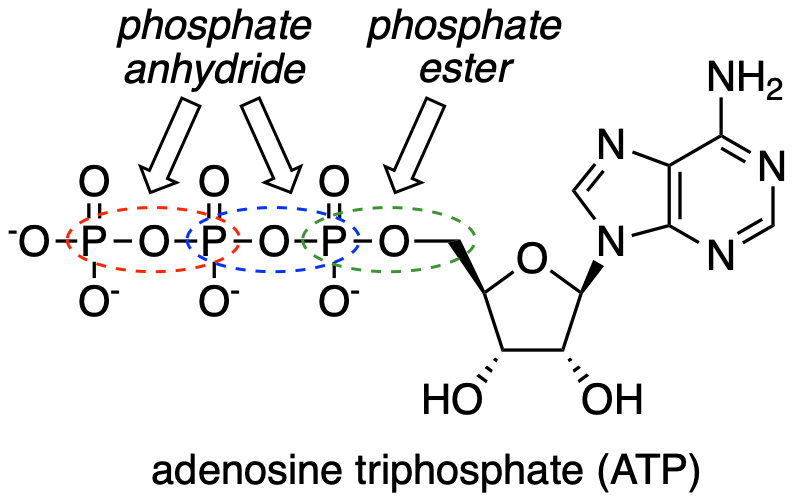
fig 3
Oxygen atoms in phosphate groups are referred to either as ‘bridging’ or ‘non-bridging’, depending on their position. An organic diphosphate has two bridging oxygens (one in the phosphate ester linkage and one in the phosphate anhydride linkage) and five non-bridging oxygens:

fig 4
A single phosphate is linked to two organic groups is called phosphate diester. The backbone of DNA is linked by phosphate diesters.
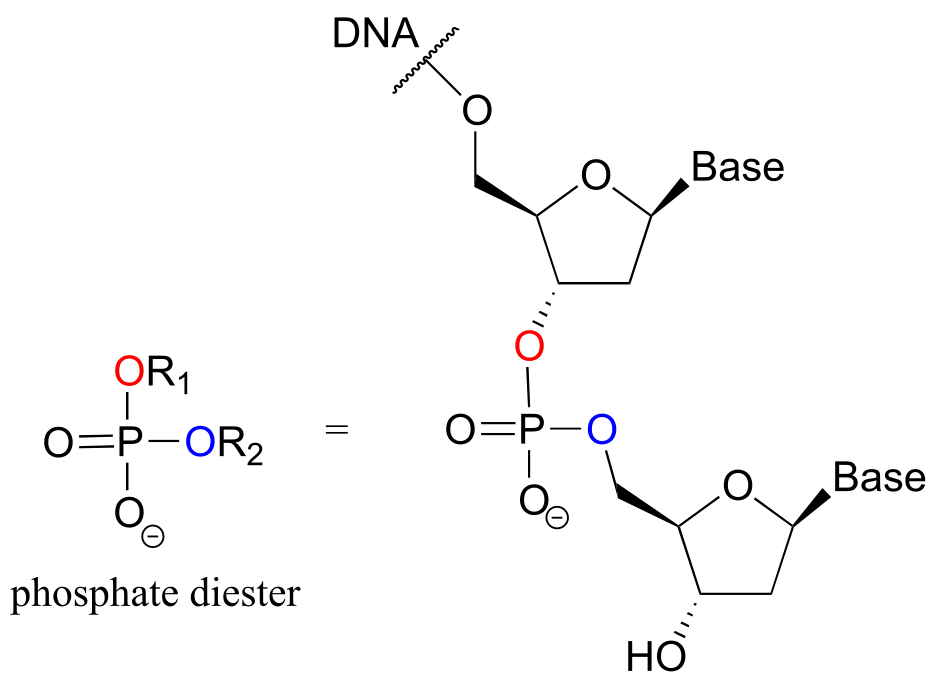
fig 5
Organic phosphates are often abbreviated using ‘OP’ and ‘OPP’ for mono- and diphosphates, respectively. For example, glucose-6-phosphate and isopentenyl diphosphate are often depicted as shown below. Notice that the ‘P’ abbreviation includes the associated oxygen atoms and negative charges.
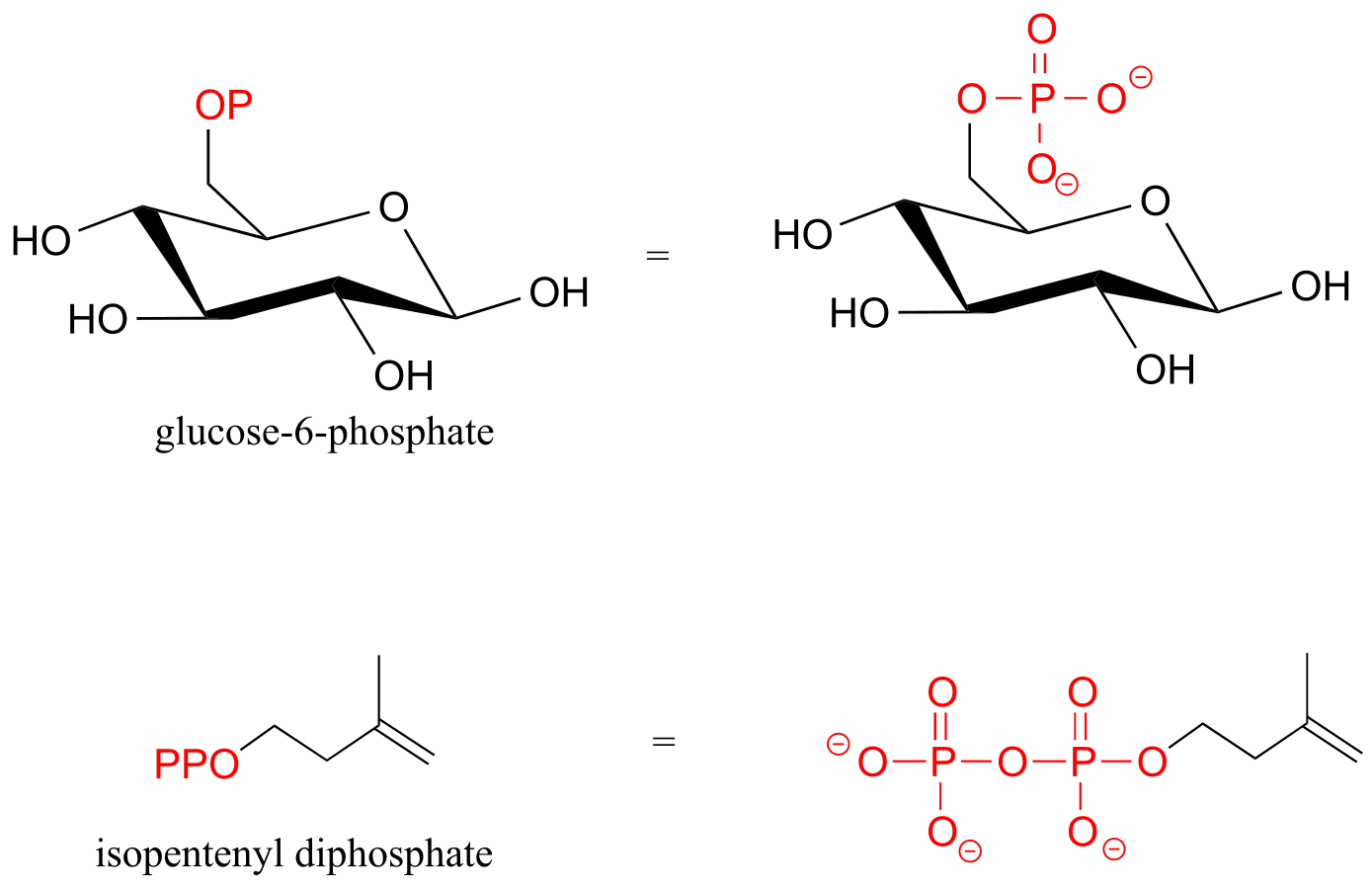
fig 6
Exercise 9.1: Consider the biological compounds below, some of which are shown with abbreviated structures:
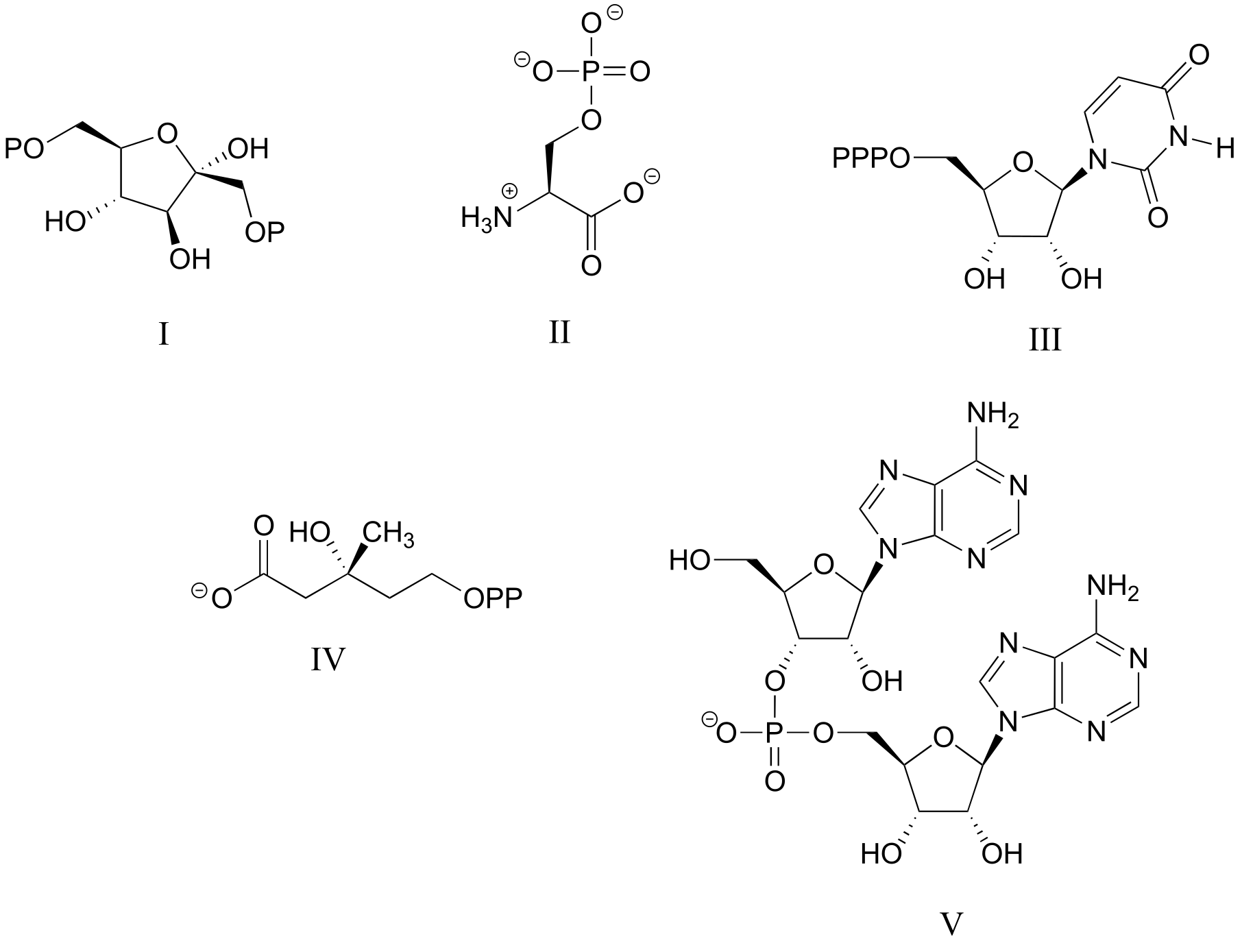
fig 4a
a) Which contain one or more phosphate anhydride linkages? Specify the number of phosphate anhydride linkages in your answers.
b) Which contain one or more phosphate monoesters? Again, specify the number for each answer.
c) Which contain a phosphate diester?
d) Which could be described as an organic diphosphate?
e) For each compound, specify the number of bridging and non-bridging oxygens in the phosphate group.
9.1B: Acid constants and protonation states#
Phosphoric acid is triprotic, meaning that it has three acidic protons available to donate, with pKa values of 1.0, 6.5, and 13.0, respectively. (da Silva and Williams)
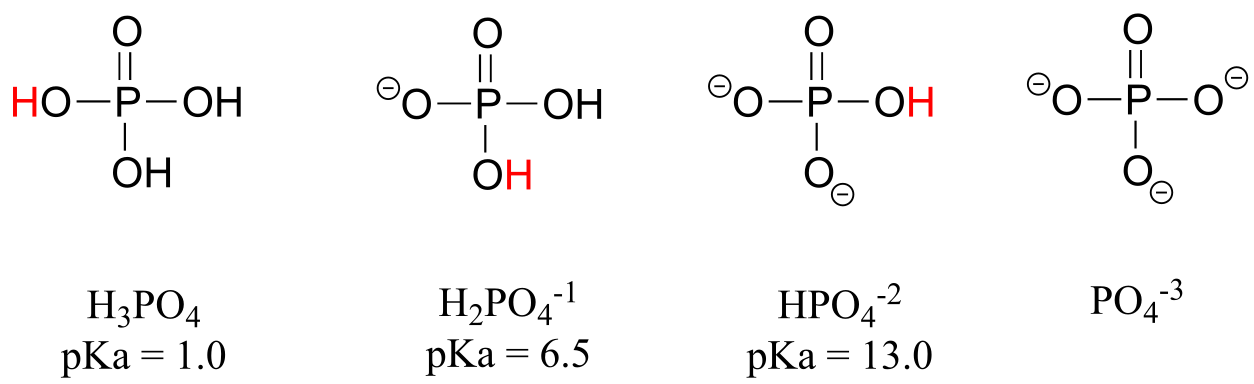
fig 7
These acid constant values, along with the Henderson-Hasselbalch equation (section 7.2C) tell us that, at the physiological pH of approximately 7, somewhat more than half of the phosphate species will be in the HPO4-2 state, and slightly less than half will be in the H2PO4-1 state, meaning that the average net charge is between -1.5 and -2.0.
Phosphate diesters have a pKa of about 1, meaning that they carry a full negative charge at physiological pH.
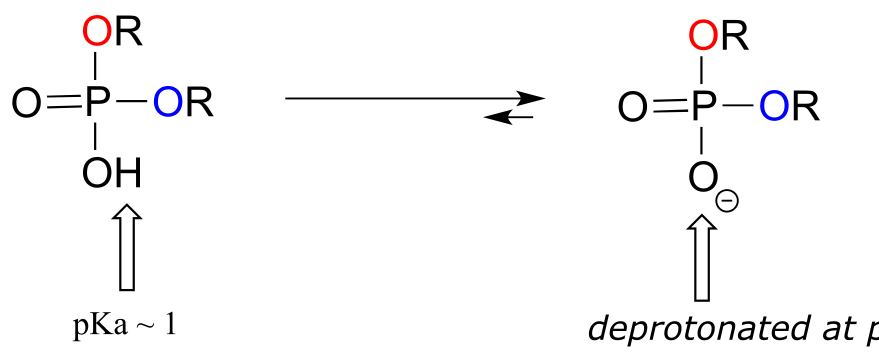
fig 7a
Organic monophosphates, diphosphates, and triphosphates all have net negative charges and are partially protonated at physiological pH, but by convention are usually drawn in the fully deprotonated state.
Exercise 9.2: Explain why the second pKa of phosphoric acid is so much higher than the first pKa.
Exercise 9.3: What is the approximate net charge of inorganic phosphate in a solution buffered to pH 1?
Recall from section 8.4 that good leaving groups in organic reactions are, as a rule, weak bases. In laboratory organic reactions, leaving groups are often halides or toluenesulfonates (section 8.4), both of which are weak bases. In biological organic reactions, phosphates are very common leaving groups. These could be inorganic phosphate, inorganic pyrophosphate, or organic monophosphates, all of which are weakly basic, especially when coordinated to metal cations such as Mg+2 in the active site of an enzyme. We will see many examples of phosphate leave groups in this and subsequent chapters.
9.1C: Bonding in phosphates#
Looking at the location of phosphorus on the periodic table, you might expect it to bond and react in a fashion similar to nitrogen, which is located just above it in the same column. Indeed, phosphines - phosphorus analogs of amines - are commonly used in the organic laboratory.
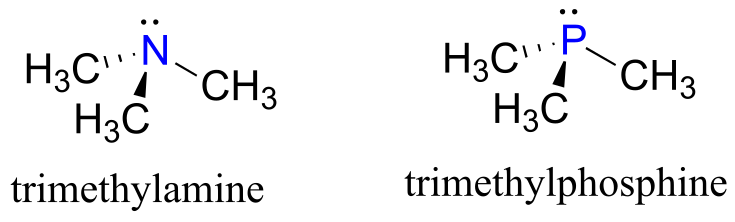
fig 8
However it is in the form of phosphate, rather than phosphine, that phosphorus plays its main role in biology.
The four oxygen substituents in phosphate groups are arranged about the central phosphorus atom with tetrahedral geometry, however there are a total of five bonds to phosphorus - four σ bonds and one delocalized π bond.
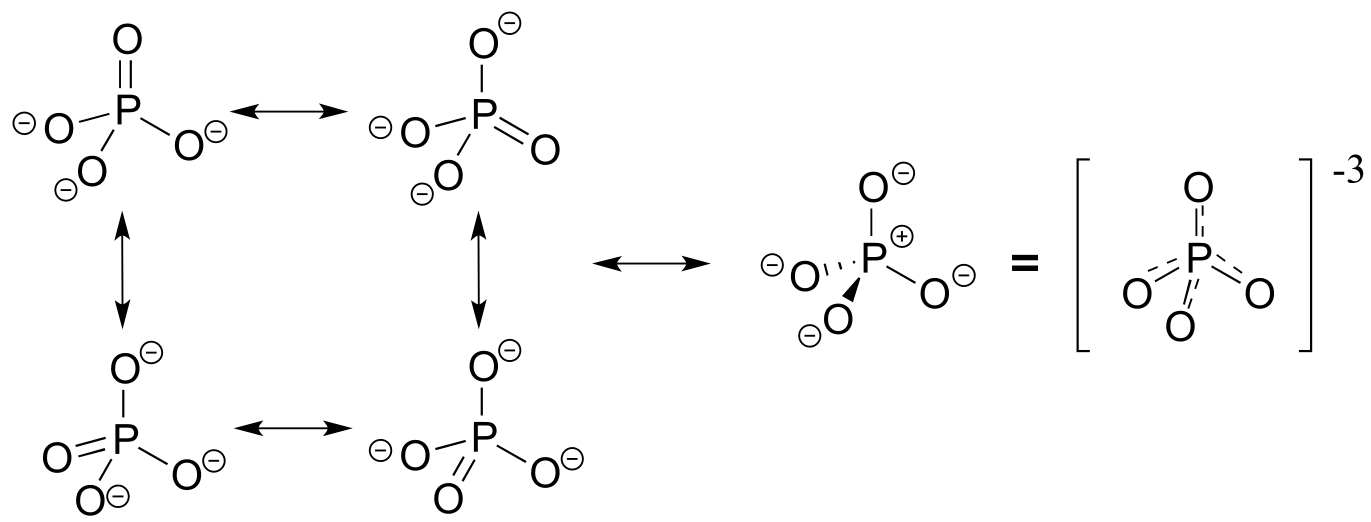
fig 9
Phosphorus can break the ‘octet rule’ because it is on the third row of the periodic table, and thus has d orbitals available for bonding. The minus 3 charge on a fully deprotonated phosphate ion is spread evenly over the four oxygen atoms, and each phosphorus-oxygen bond can be considered to have 25% double bond character: in other words, the bond order is 1.25.
Recall from section 2.1 the hybrid bonding picture for the tetrahedral nitrogen in an amine group: a single 2s and three 2p orbitals combine to form four sp3 hybrid orbitals, three of which form σ bonds and one of which holds a lone pair of electrons.
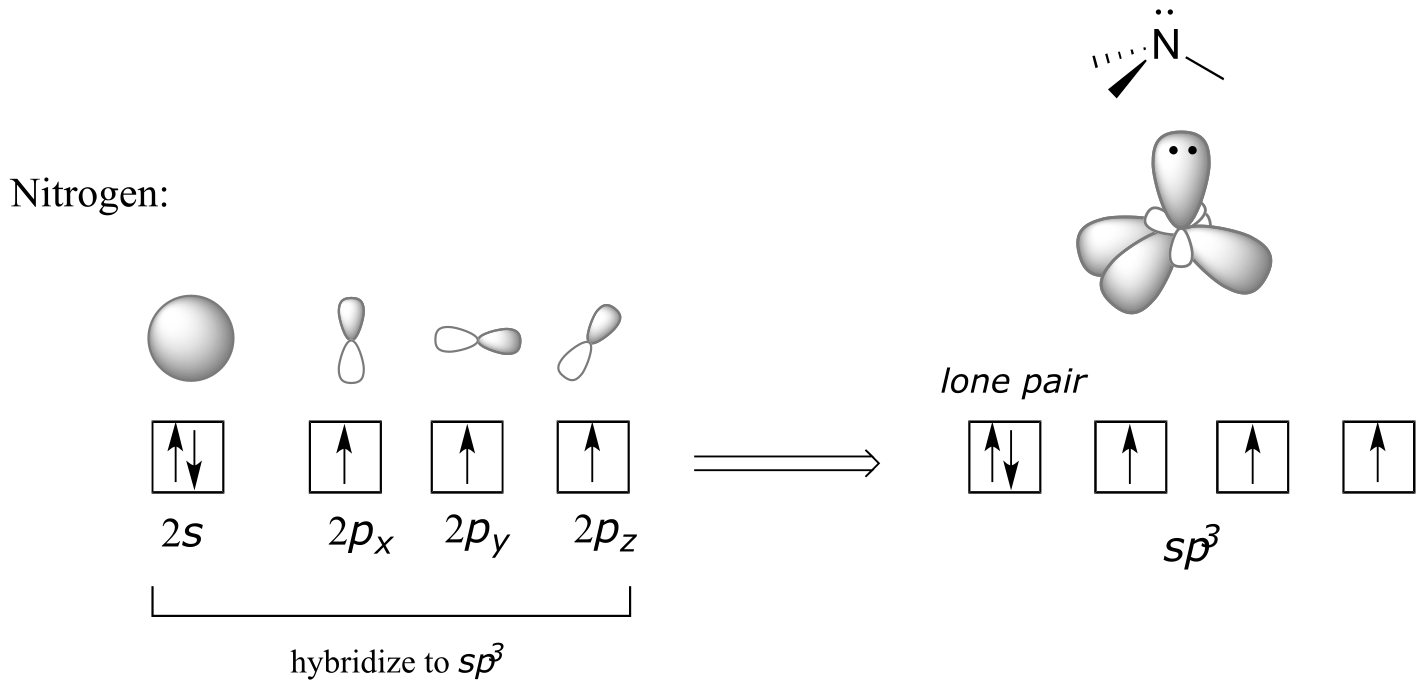
fig 10
In the hybrid orbital picture for phosphate ion, a single 3s and three 3p orbitals also combine to form four sp3 hybrid orbitals with tetrahedral geometry. In contrast to an amine, however, four of the five valance electrons on phosphorus occupy sp3 orbitals, and the fifth occupies an unhybridized 3d orbital.

fig 11
This orbital arrangement allows for four σ bonds with tetrahedral geometry in addition to a fifth, delocalized π bond formed by π overlap between the half-filled 3d orbital on phosphorus and 2p orbitals on the oxygen atoms.
In phosphate esters, diesters, and anhydrides the π bonding is delocalized primarily over the non-bridging bonds, while the bridging bonds have mainly single-bond character. In a phosphate diester, for example, the two non-bridging oxygens share a -1 charge, as illustrated by the two major resonance contributors below. The bonding order for the bridging P-O bonds in a phosphate diester group is about 1, and for the non-bridging P-O bonds about 1.5. In the resonance contributors in which the bridging oxygens are shown as double bonds (to the right in the figure below), there is an additional separation of charge - thus these contributors are minor and make a relatively unimportant contribution to the overall bonding picture.
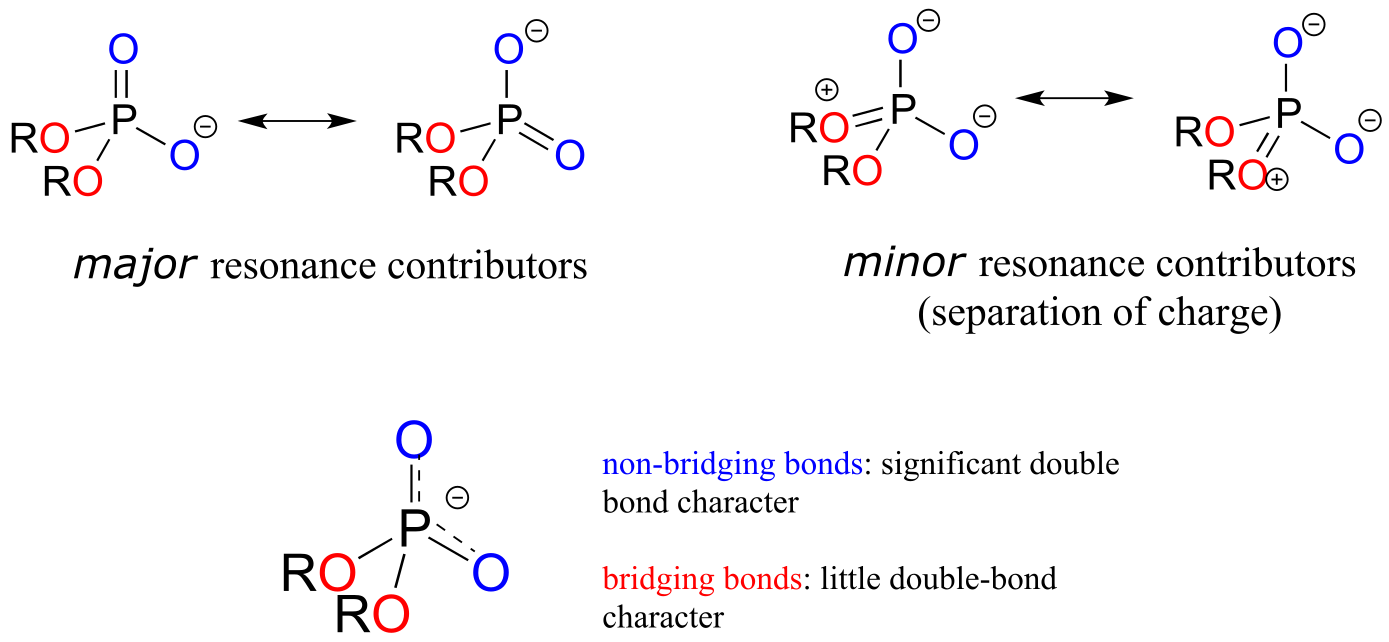
fig 12
Exercise 9.4: Draw all of the resonance structures showing the delocalization of charge on a (fully deprotonated) organic monophosphate. If a ‘bond order’ of 1.0 is a single bond, and a bond order of 2.0 is a double bond, what is the approximate bond order of bridging and non-bridging P-O bonds?
Throughout this book, phosphate groups will often be drawn without attempting to show tetrahedral geometry, and π bonds and negative charges will usually be shown localized to a single oxygen. This is done for the sake of simplification - however it is important always to remember that the phosphate group is really tetrahedral, the negative charges are delocalized over the non-bridging oxygens, and that there is some degree of protonation at physiological pH (with the exception of the phosphate diester group).
9.2: Phosphate transfer reactions - an overview#
In a phosphate transfer reaction, a phosphate group is transferred from a phosphate group donor molecule to a phosphate group acceptor molecule:
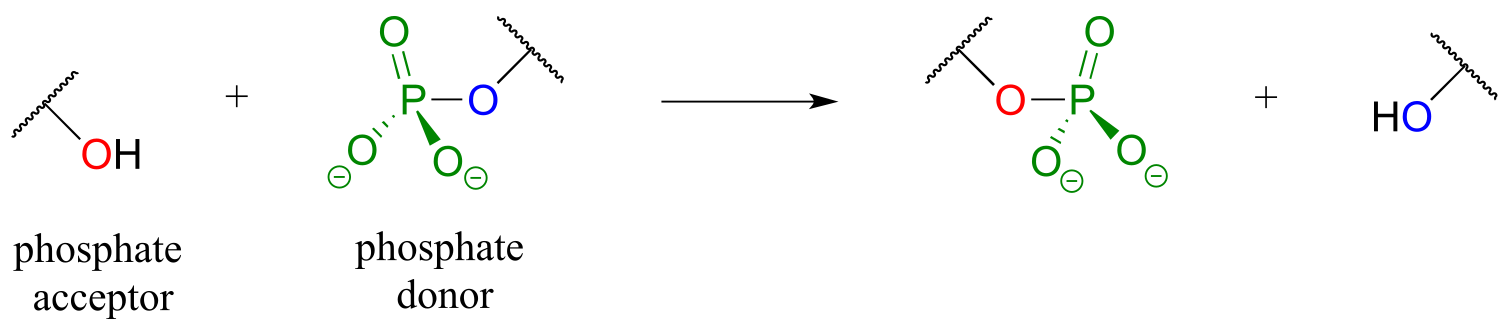
fig 13
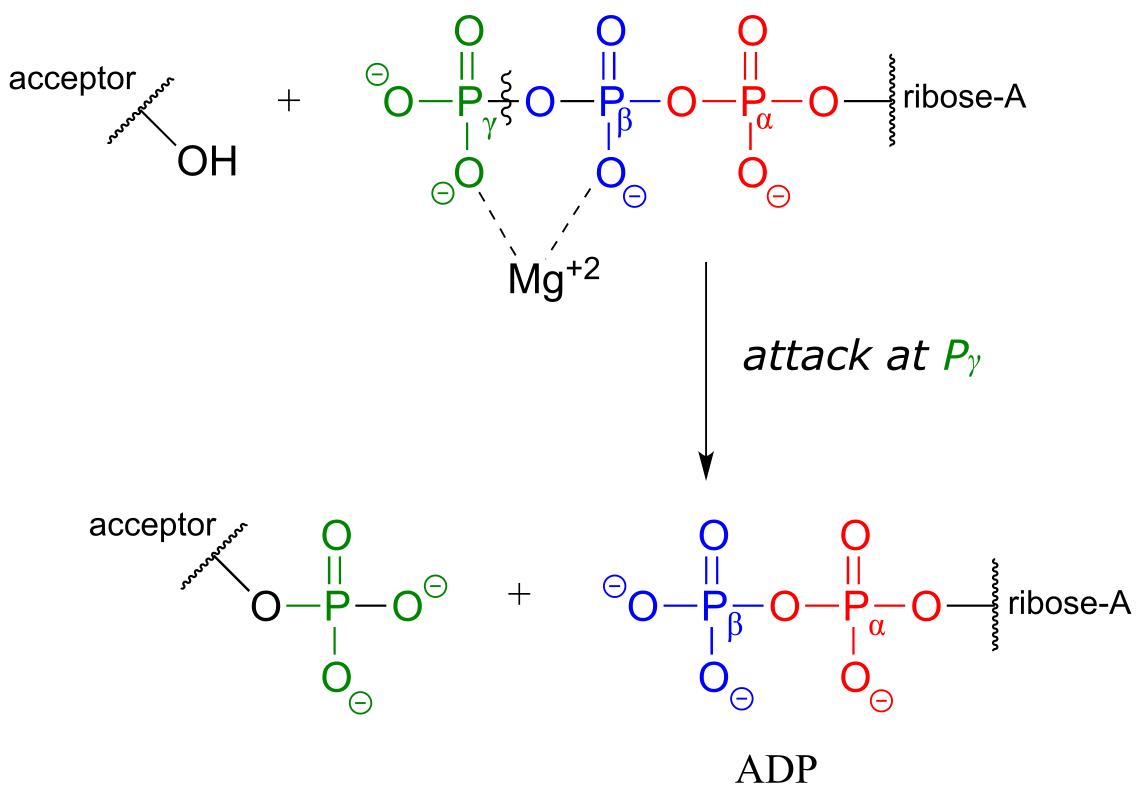
A very important aspect of biological phosphate transfer reactions is that the electrophilicity of the phosphorus atom is usually enhanced by the Lewis acid (electron-accepting) effect of one or more magnesium ions. Phosphate transfer enzymes generally contain a Mg2+ ion bound in the active site in a position where it can interact with non-bridging phosphate oxygens on the substrate. The magnesium ion pulls electron density away from the phosphorus atom, making it more electrophilic.

fig 17
Without this metal ion interaction, a phosphate is actually a poor electrophile, as the negatively-charged oxygens shield the phosphorus center from attack by a nucleophile.
Note: For the sake of simplicity and clarity, we will not draw the magnesium ion or other active site groups interacting with phosphate oxygens in most figures in this chapter - but it is important to keep in mind that these interactions play an integral role in phosphate transfer reactions.
Mechanistically speaking, a phosphate transfer reaction at a phosphorus center can be though of as much like a SN2 reaction at a carbon center. Just like in an SN2 reaction, the nucleophile in a phosphoryl transfer approaches the electrophilic center from the backside, opposite the leaving group:
**
**
Concerted model:
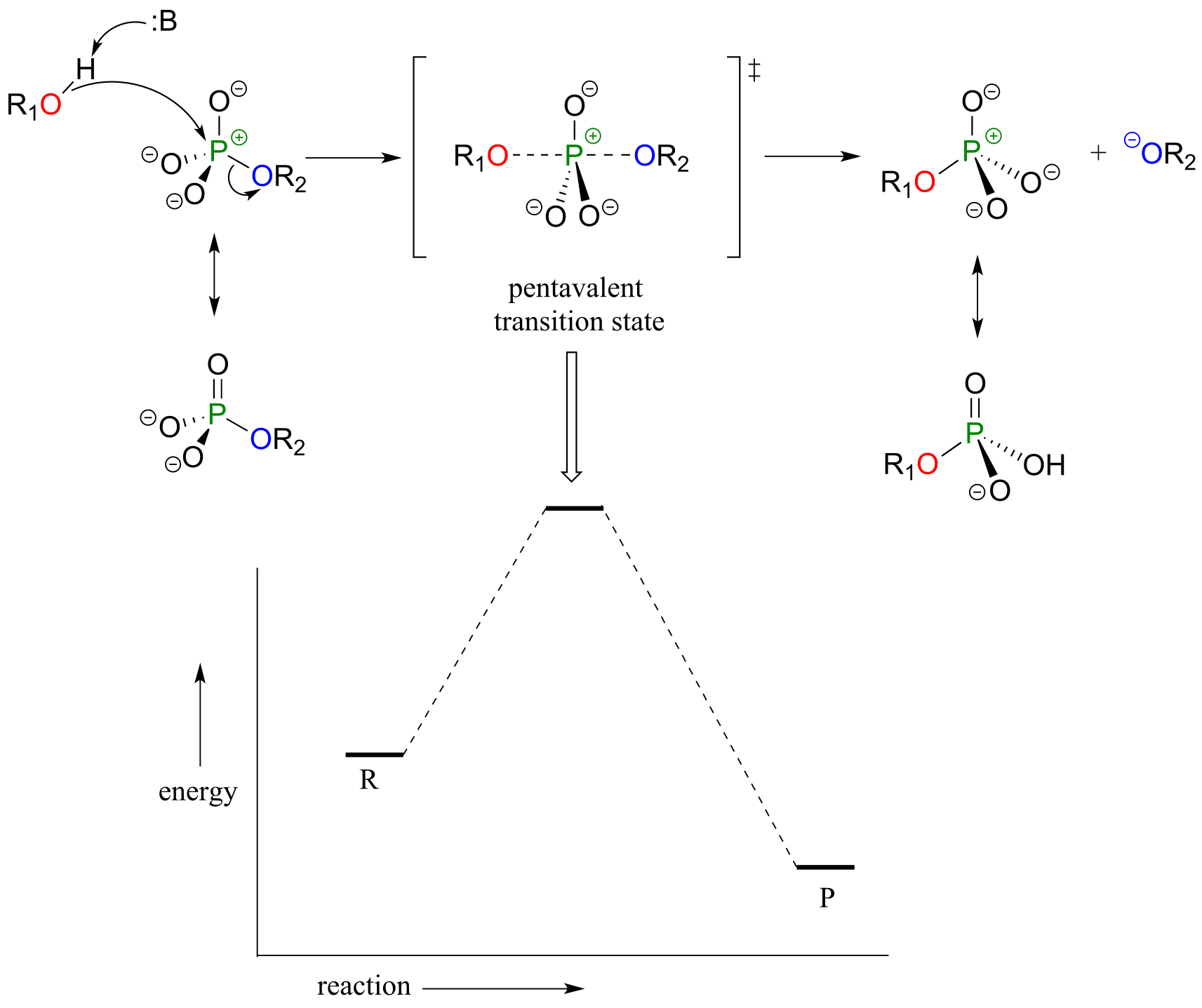
fig 17a
As the nucleophile gets closer and the leaving group begins its departure, the bonding geometry at the phosphorus atom changes from tetrahedral to trigonal bipyramidal at the pentavalent (5-bond) transition state. As the phosphorus-nucleophile bond gets shorter and the phosphorus-leaving group bond grows longer, the bonding picture around the phosphorus atom returns to its original tetrahedral state, but the stereochemical configuration has been ‘flipped’, or inverted.
In the trigonal bipyramidal transition state, the five substituents are not equivalent: the three non-bridging oxygens are said to be equatorial (forming the base of a trigonal bipyramid), while the nucleophile and the leaving group are said to be apical (occupying the tips of the two pyramids).
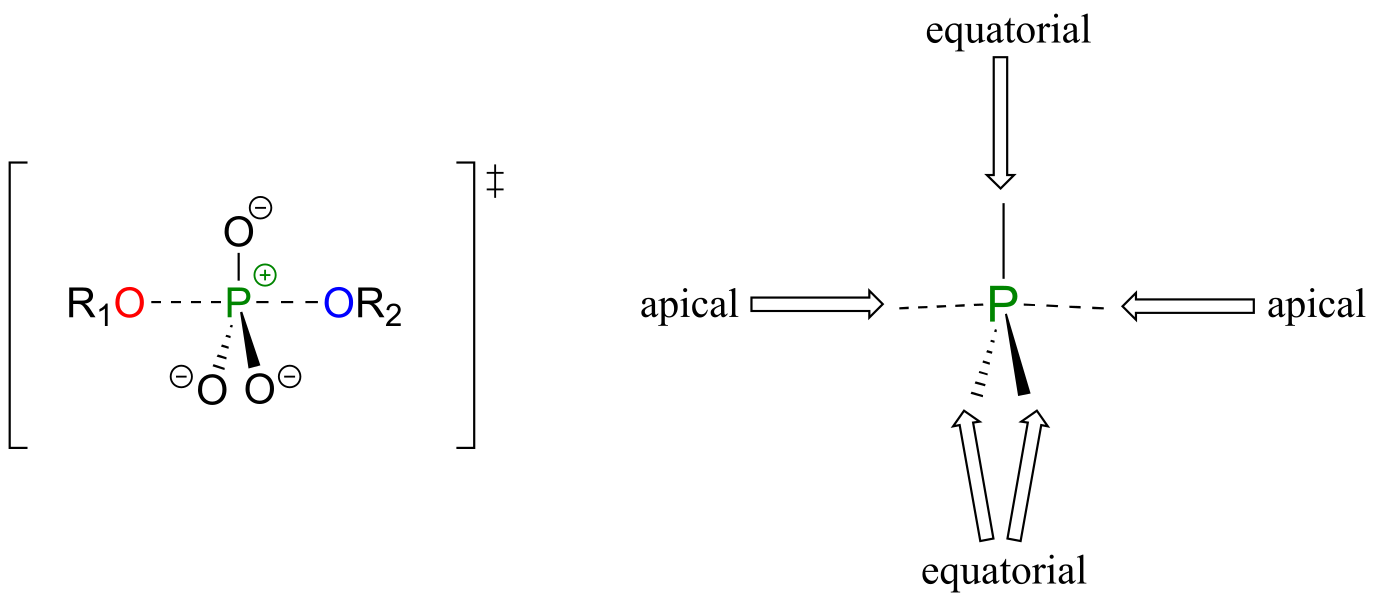
fig 17b
Although stereochemical inversion in phosphoryl transfer reactions is predicted by theory, the fact that phosphoryl groups are achiral made it impossible to observe the phenomenon directly until 1978, when a group of researchers was able to synthesize organic phosphate esters in which stable oxygen isotopes 17O and 18O were specifically incorporated. This created a chiral phosphate center.
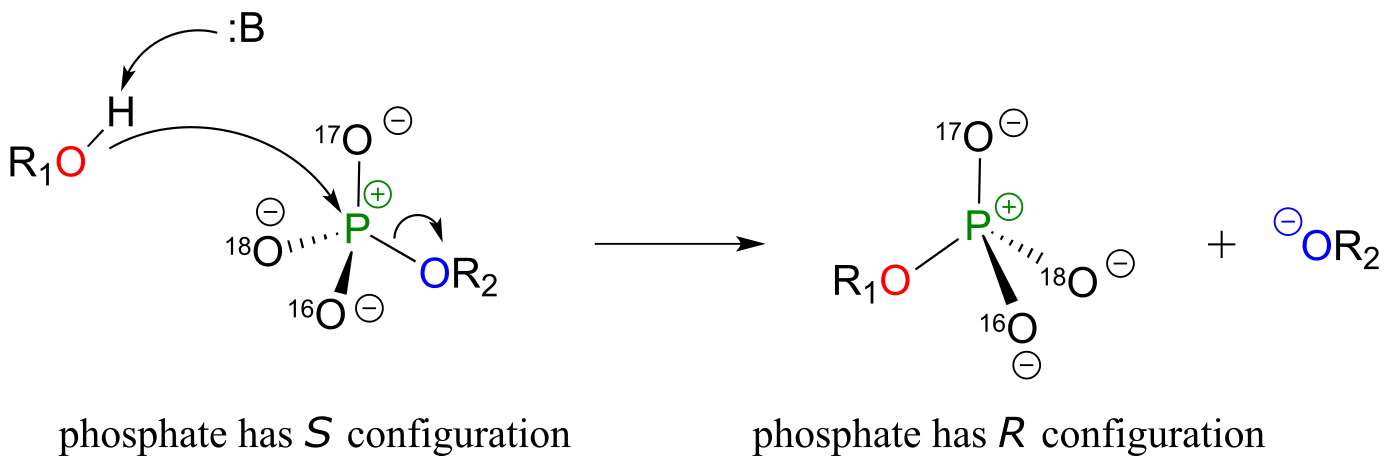
fig 17c
Subsequent experiments with phosphoryl transfer-catalyzing enzymes confirmed that these reactions proceed with stereochemical inversion. (Nature 1978 275, 564; Ann Rev Biochem 1980 49, 877).
The concerted (SN2-like) is not the only mechanism that has been proposed for these reactions - in fact, two other possible mechanisms have been suggested. In an alternative two-step mechanistic model, the nucleophile could attack first, forming
a pentavalent, trigonal bipyramidal intermediate (as apposed to a pentavalent transition state). The reaction is completed when the leaving group is expelled. The intermediate species would occupy an energy valley between the two transition states.
**
**
Addition-elimination model:
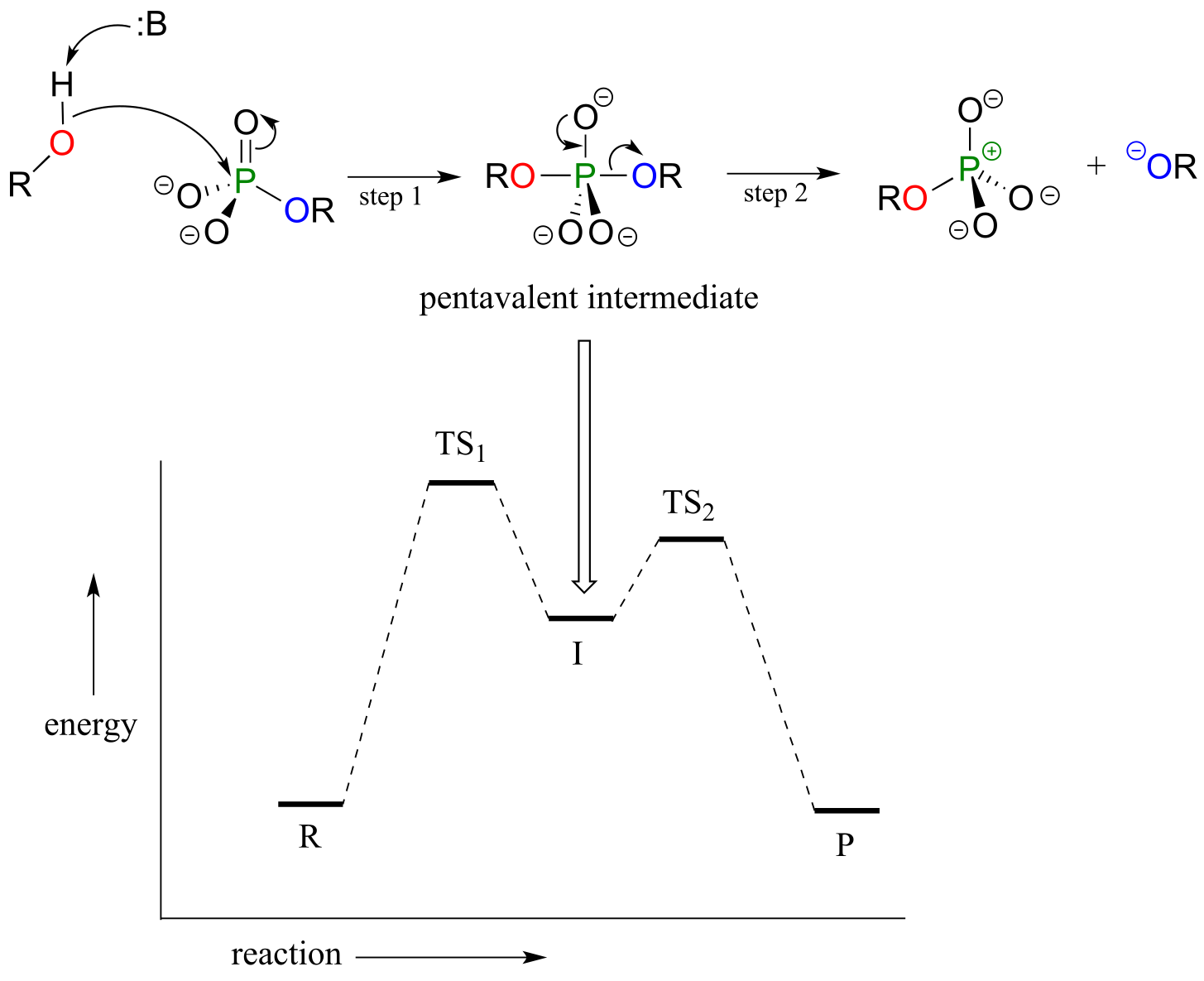
fig 17e
This is often referred to as an ‘addition-elimination’ mechanism - the nucleophile adds to the phosphate first, forming a pentavalent intermediate, and then the leaving group is eliminated.
An addition-elimination mechanism with a pentavalent intermediate is not possible for an SN2 reaction at a carbon center, because carbon, as a second-row element, does not have any d orbitals and cannot form five bonds. Phosphorus, on the other hand, is a third-row element and is quite capable of forming more than four bonds. Phosphorus pentachloride, after all, is a stable compound that has five bonds to chlorine arranged in trigonal bipyramidal geometry around the central phosphorus.
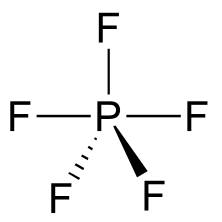
fig 17f
The phosphorus atom in PCl5 (and in the hypothetical pentavalent intermediate pictured above) is considered to be sp3d hybridized:
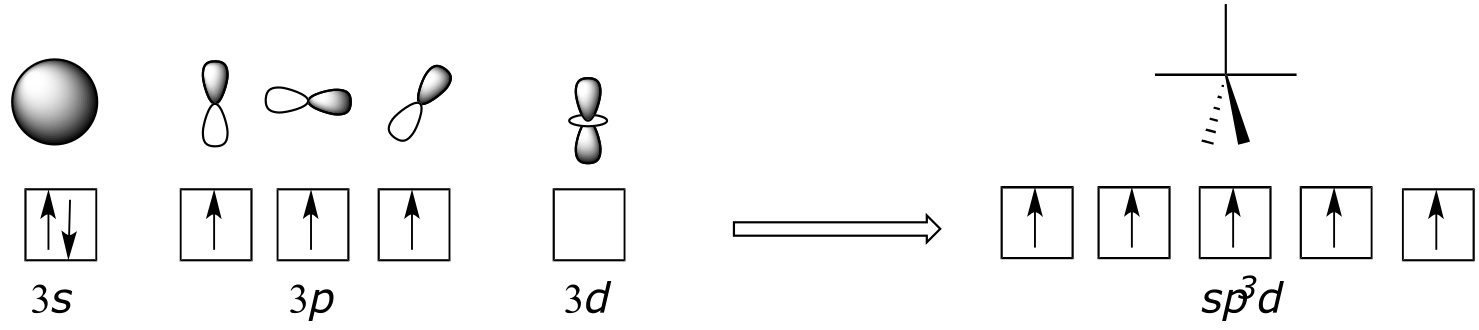
fig 17g
There is a third possibility: the reaction could proceed in an SN1-like manner: in other words, elimination-addition. In this model, the phosphorus-leaving group bond breaks first, resulting in a metaphosphate intermediate. This intermediate, which corresponds to the carbocation intermediate in an SN1 reaction and like a carbocation has trigonal planar geometry, is then attacked by the nucleophile to form the reaction product.
Elimination-addition model:
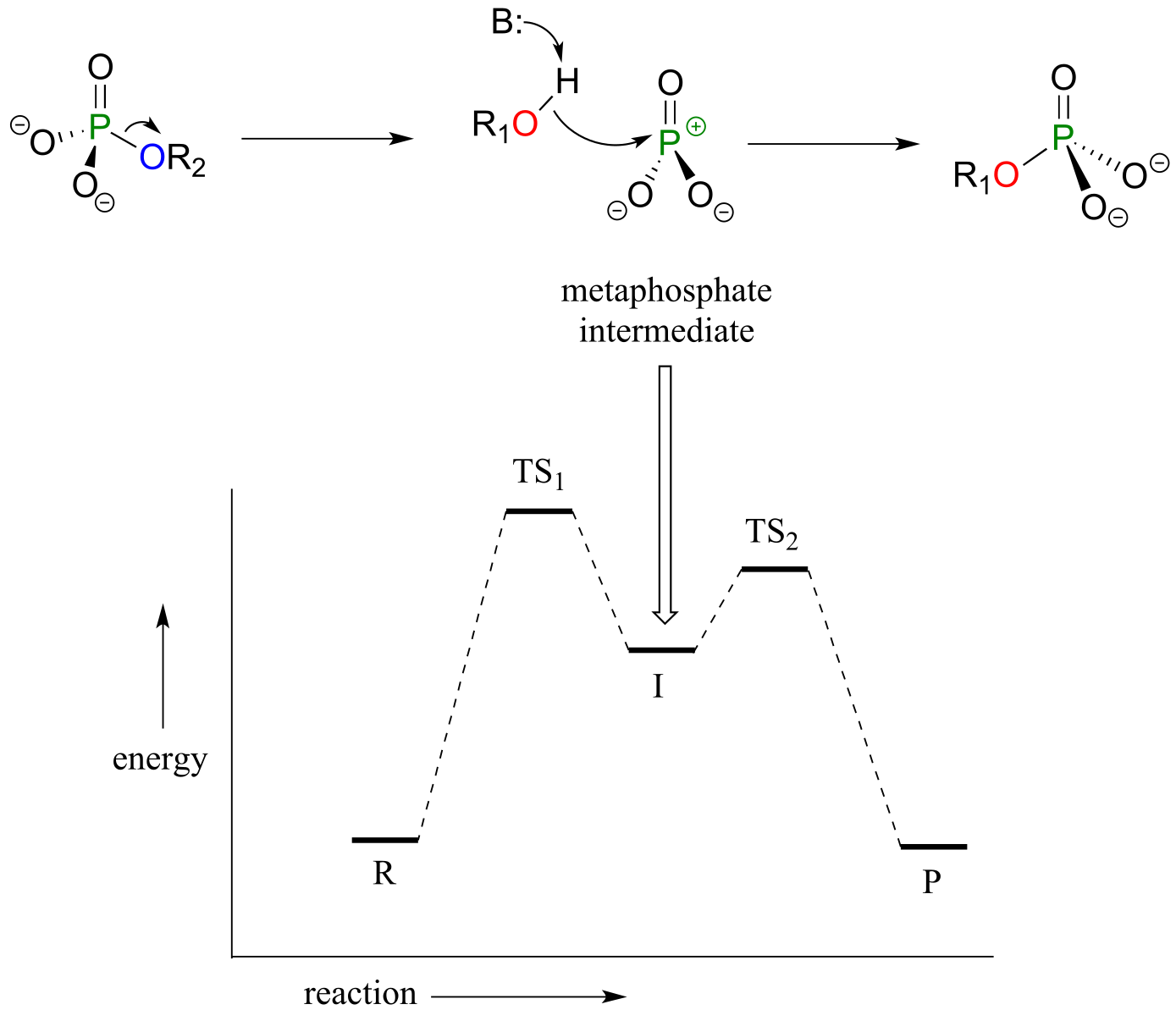
fig 5
fig 17h
So which mechanistic model - concerted (SN2-like), addition-elimination, or elimination-addition - best describes enzymatic phosphate transfer reactions? Chemists love to investigate and debate questions like this! Just like with the SN1/SN2 argument discussed in chapter 8, it really boils down to one question: which happens first, bond-forming or bond-breaking - or do these two events occur at the same time? From the evidence accumulated to date, it appears that many enzymatic phosphate transfer reactions can best be described by the concerted model, although there is still argument about this, and still many unanswered questions about other details of how these reactions are catalyzed in active sites. Considering the importance of phosphate transfer reactions in metabolic pathways, this area is clearly a very promising one for further investigation. If you are interesting in learning more about this research, a great place to start is a review article written by Professor Daniel Herschlag at Stanford University (Annu. Rev. Biochem. 2011, 80, 669).
For the sake of simplicity and clarity, phosphoryl transfers in this text will be depicted using the concerted model.
Exercise 9.5: Predict the approximate angles between the two bonds indicated in a phosphate transfer transition state. Oa refers to an oxygen at the apical position, and Oe to an oxygen in the equatorial position.a) Oa-P-Oa b) Oa-P-Oe c)Oe-P-Oe
9.3: ATP, the principal phosphate donor#
Thus far we have been very general in our discussion of phosphate transfer reactions, referring only to generic ‘donor’ and ‘acceptor’ species. It’s time to get more specific. The most important donor of phosphate groups in the cell is a molecule called adenosine triphosphate, commonly known by its abbreviation ATP.
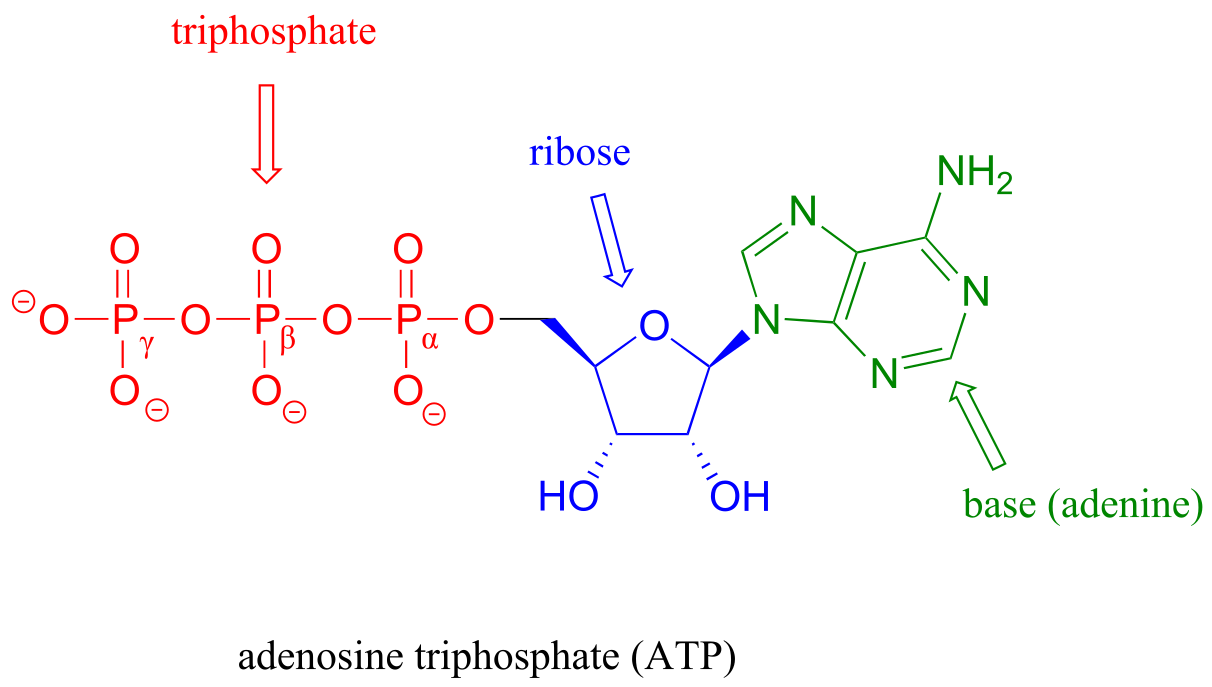
fig 21
Notice that there are essentially three parts to the ATP molecule: an adenine nucleoside ‘base’, a five-carbon sugar (ribose), and triphosphate. The three phosphates are designated by Greek letters α, β, and γ, with the α phosphate being the one closest to the ribose. Adenosine diphosphate (ADP) and adenosine monophosphate (AMP) are also important players in the reactions of this chapter.
ATP is a big molecule, but the bond-breaking and bond-forming events we will be studying in this chapter all happen in the phosphate part of the molecule. You will see structural drawings of ATP, ADP, and AMP abbreviated in many different ways in this text and throughout the biochemical literature, depending on what is being illustrated. For example, the three structures below are all abbreviated depictions of ATP:
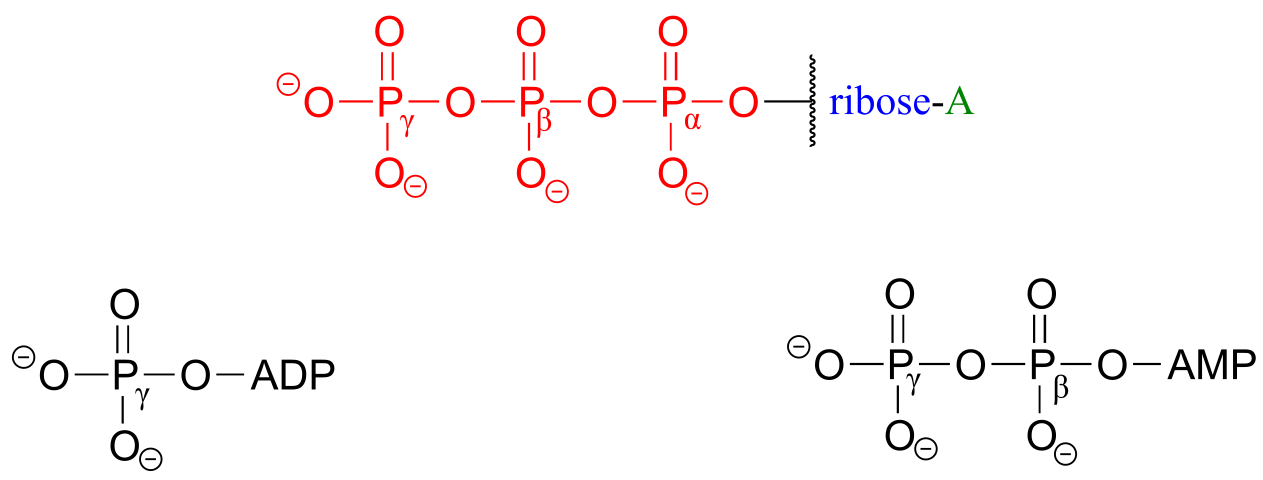
fig 22
The following exercise will give you some practice in recognizing different abbreviations for ATP and other biological molecules that contain phosphate groups.
Exercise 9.6 : Below are a number of representations, labeled A-S, of molecules that contain phosphate groups. Different abbreviations are used. Arrange A-S into groups of drawings that depict the same species (for example, group together all of the abbreviations which depict ATP).
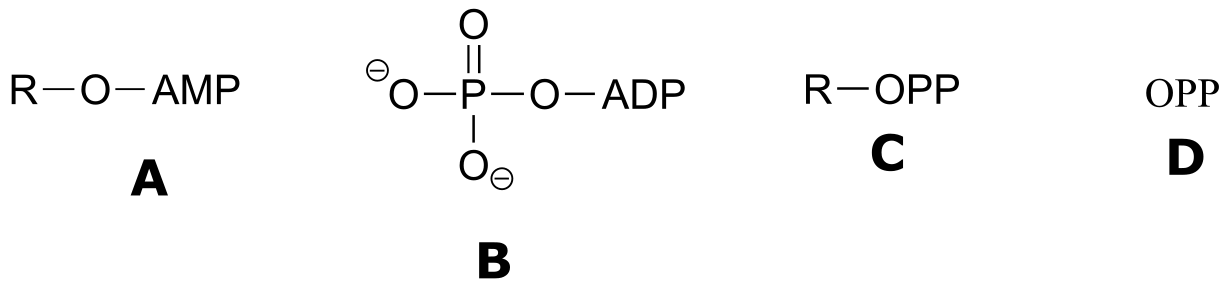
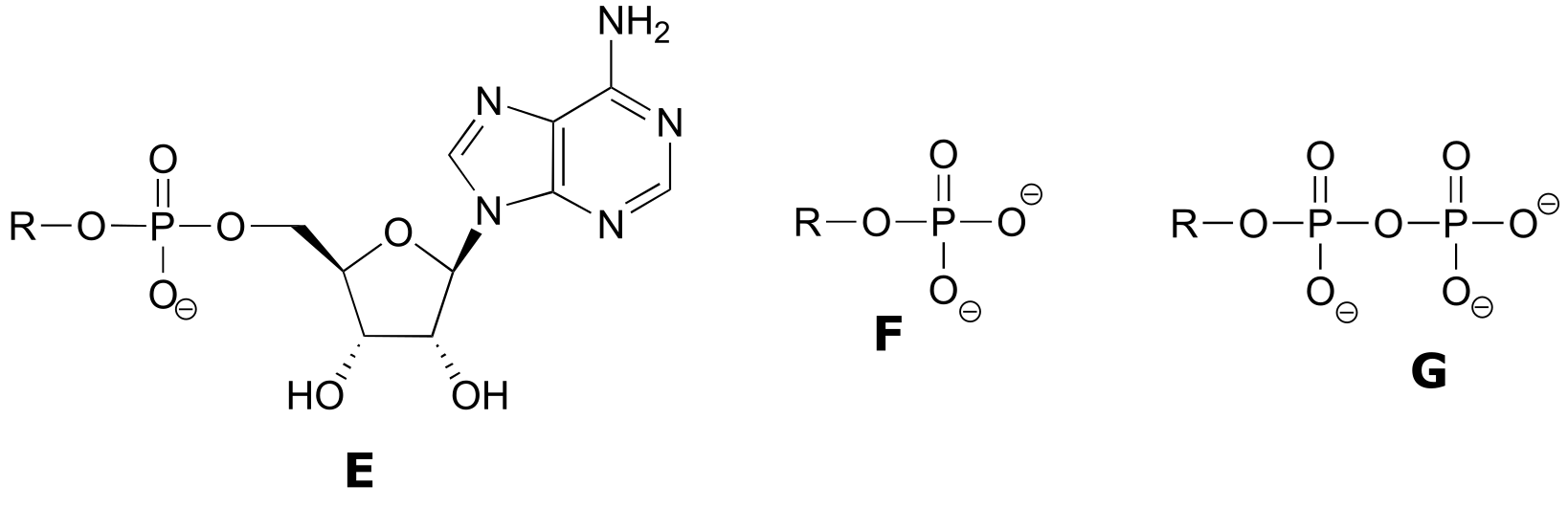
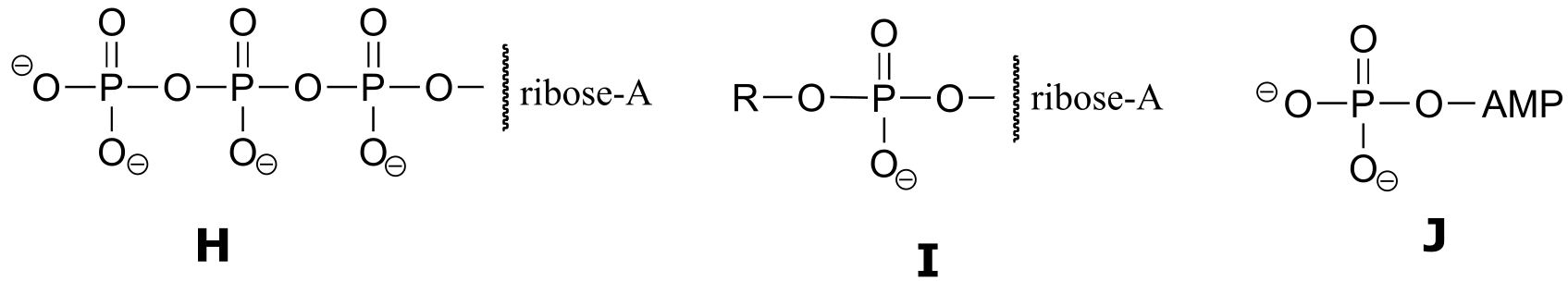
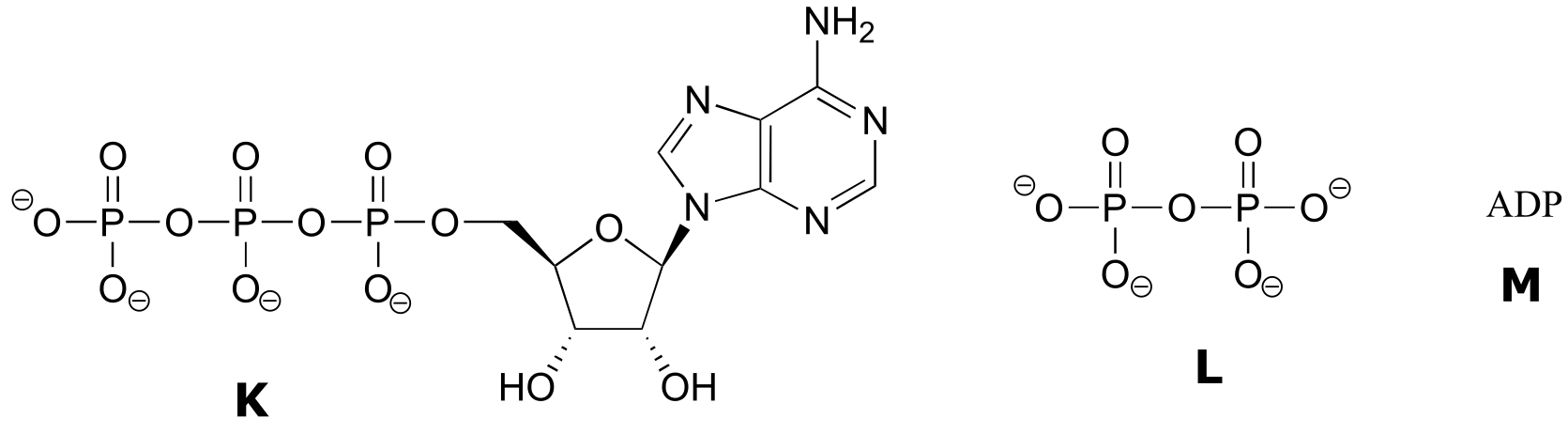

You are probably familiar with the physiological role of ATP from your biology classes - it is commonly called ‘the energy currency of the cell’. What this means is that ATP stores energy we get from the oxidation of fuel molecules such as carbohydrates or fats. The energy in ATP is stored in the two high-energy phosphate anhydride linkages.
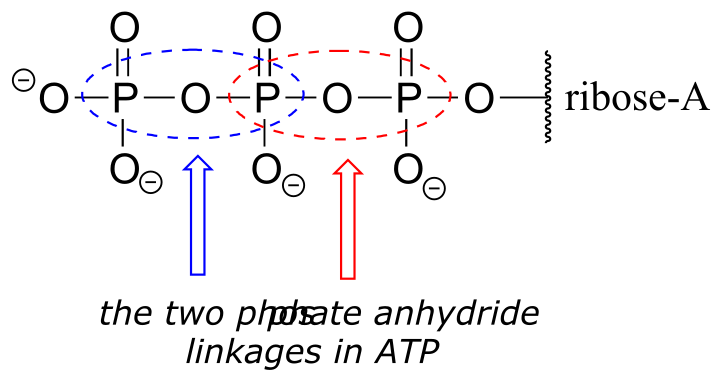
fig 23
When one or both of these phosphate anhydride links are broken as a phosphate group is transferred to an acceptor, a substantial amount of energy is released. The negative charges on the phosphate groups are separated, eliminating some of electrostatic repulsion that existed in ATP. One way to picture this is as a coil springing open, releasing potential energy.
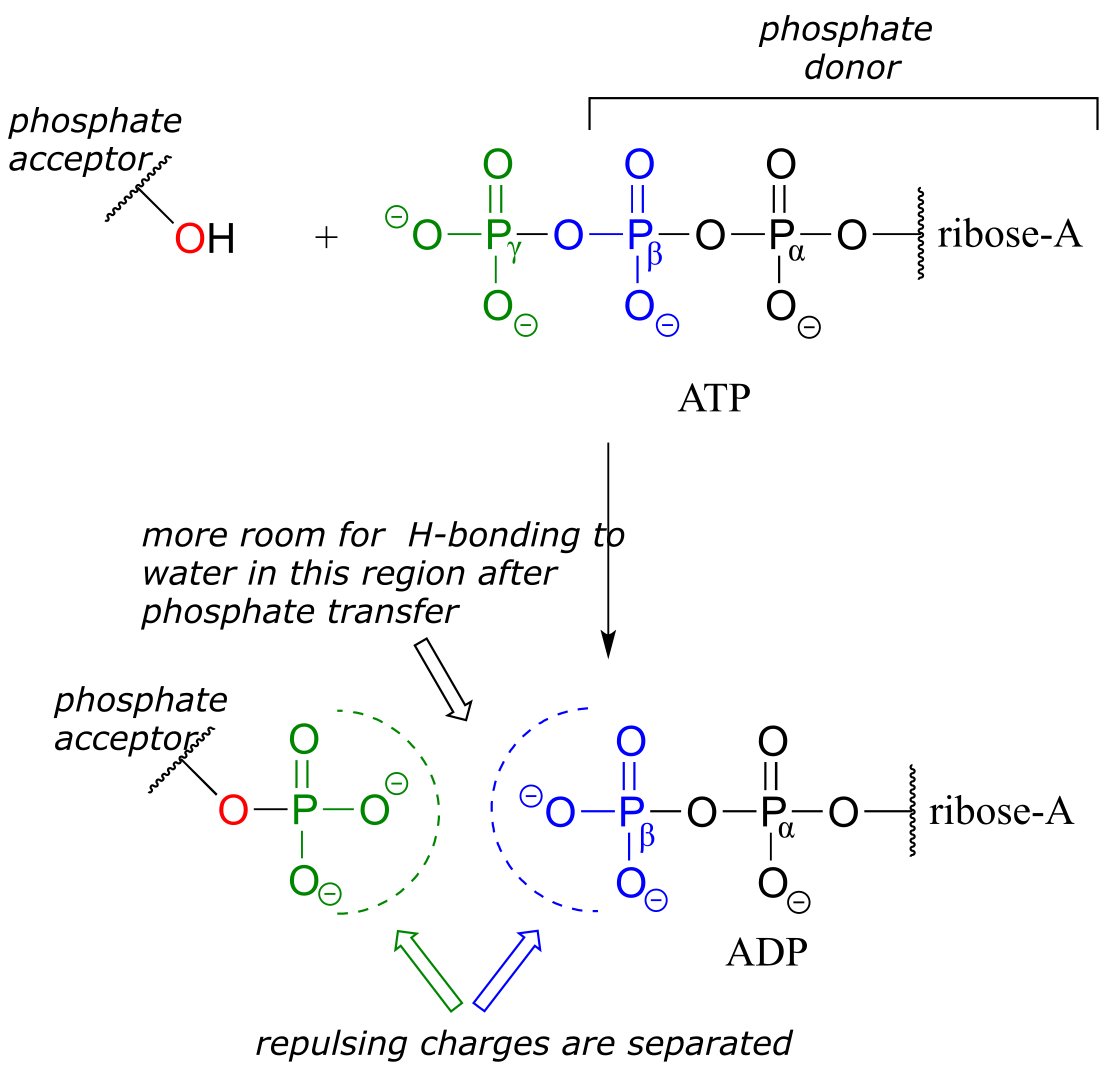
fig 24
In addition, cleavage of a phosphate anhydride bond means that surrounding water molecules are able to form more stabilizing hydrogen-bonding interactions with the products than was possible with the starting materials, again making the reaction more ‘downhill’, or exergonic.
It is important to understand that while the phosphate anhydride bonds in ATP are thermodynamically unstable (they contain a great deal of chemical energy), they are at the same time kinetically stable: ATP-cleaving reactions are exothermic, but also have a high energy barrier, making them very slow unless catalyzed by an enzyme. In other words, the release of the energy contained in ATP is highly energetic but also subject to tight control by the interaction of highly evolved enzymes in our metabolic pathways.
ATP is a versatile phosphate group donor: depending on the site of nucleophilic attack (at the α, β, or γ phosphorus), different phosphate transfer outcomes are possible. Below are the three most common patterns seen in the central metabolic pathways. A ‘squigly’ line in each figure indicates the P-O bond being broken. We will study specific examples of each of these in the coming sections.
Attack at the γ-phosphate:
Attack at the β-phosphate:
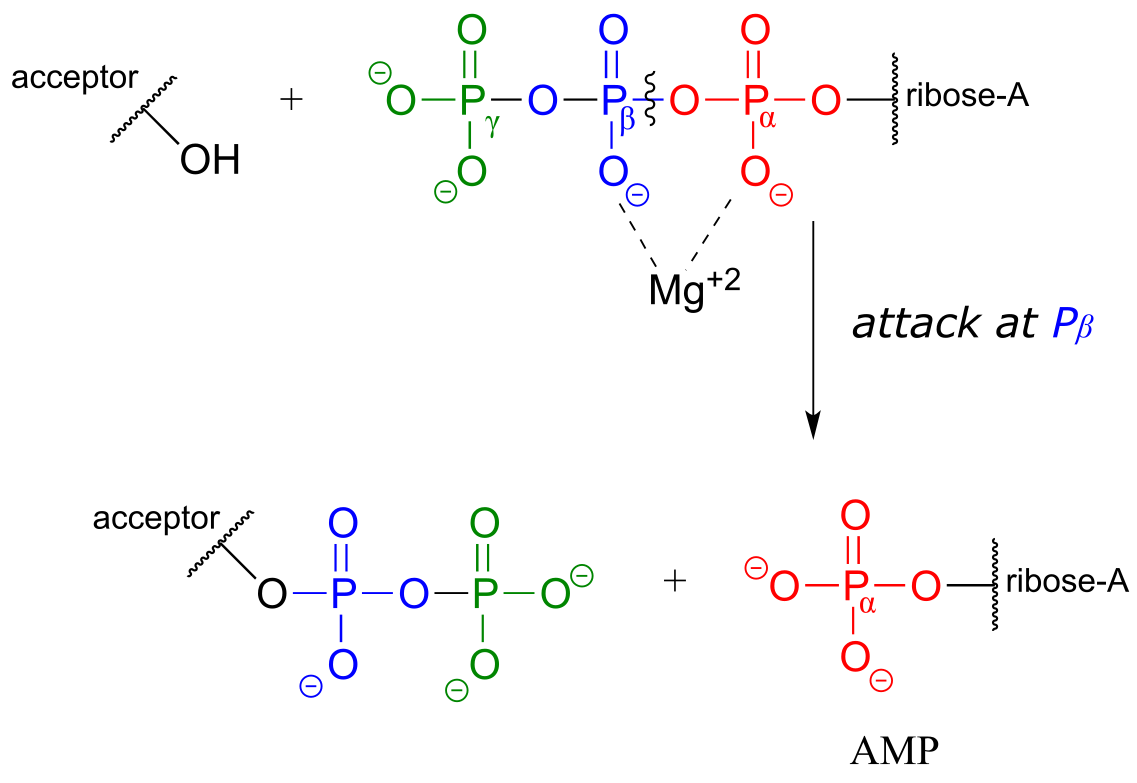
Attack at the α-phosphate:
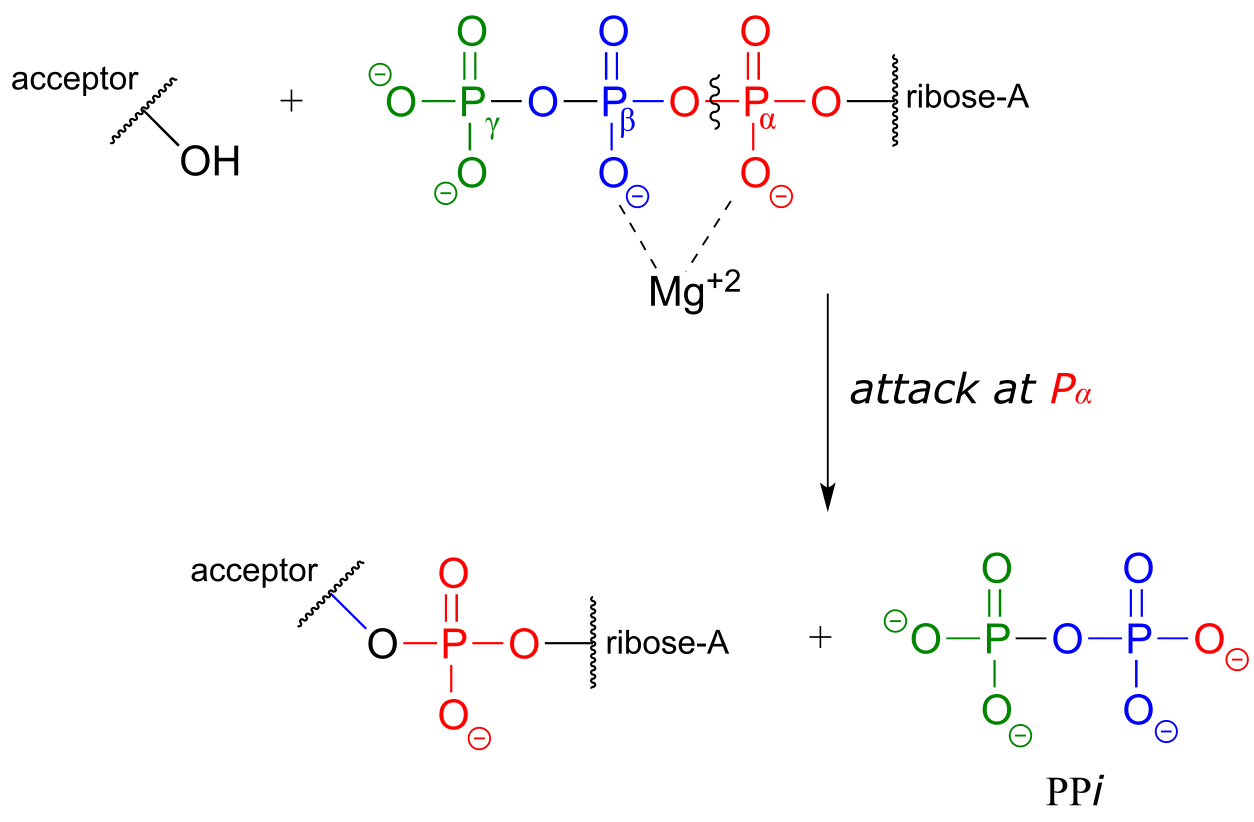
fig 24c
The common thread running through all of the ATP-dependent reactions we will see in this section is the idea that the phosphate acceptor molecule is undergoing a thermodynamically ‘uphill’ transformation to become a more reactive species. The energy for this uphill transformation comes from breaking a high-energy phosphate anhydride bond in ATP. That is why ATP is often referred to as ‘energy currency’: the energy in its anhydride bonds is used to ‘pay for’ a thermodynamically uphill chemical step.
Exercise 9.7: Propose a fourth hypothetical phosphate transfer reaction between ATP and the generic acceptor molecule in the figure above, in which inorganic phosphate (Pi) is a by-product.
Exercise 9.8 : Why is this hypothetical phosphate transfer reaction less energetically favorable compared to all of the possible ATP-cleaving reactions shown in the figure above?
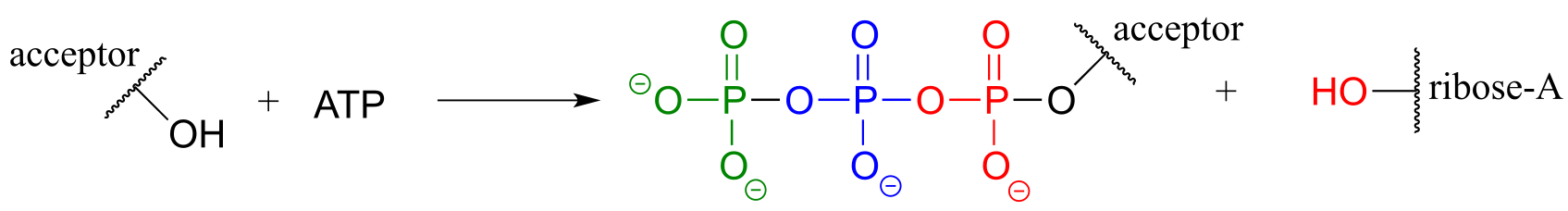
fig 24d
**
**
9.4: Phosporylation of alcohols#
A broad family of enzymes called kinases catalyze transfer of a phosphate group from ATP to an alcohol acceptor. Mechanistically, the alcohol oxygen acts as a nucleophile, attacking the electrophilic γ-phosphorus of ATP and expelling ADP.
Glucose is phosphorylated in the first step of the glycolysis pathway by the enzyme hexose kinase (EC 2.7.1.1), forming glucose-6-phosphate.
Hexose kinase mechanism
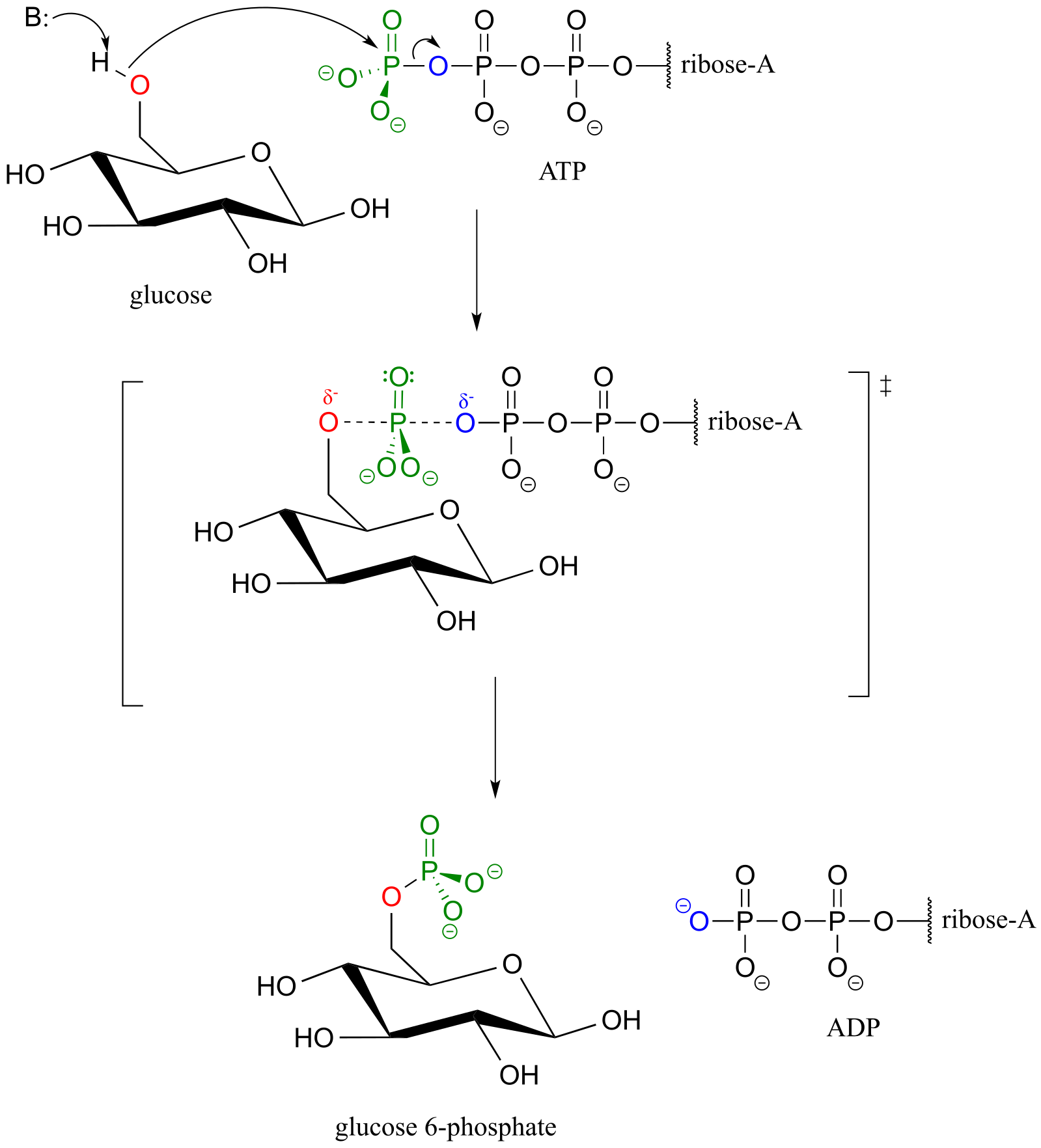
fig 26
Here is a shorthand way to depict this reaction. Notice the “ATP in, ADP out” notation used below, indicating that ATP is one of the reactants and ADP is one of the products. From here on, we will frequently use this common convention to indicate reaction participants whose structures are not drawn out in a figure.
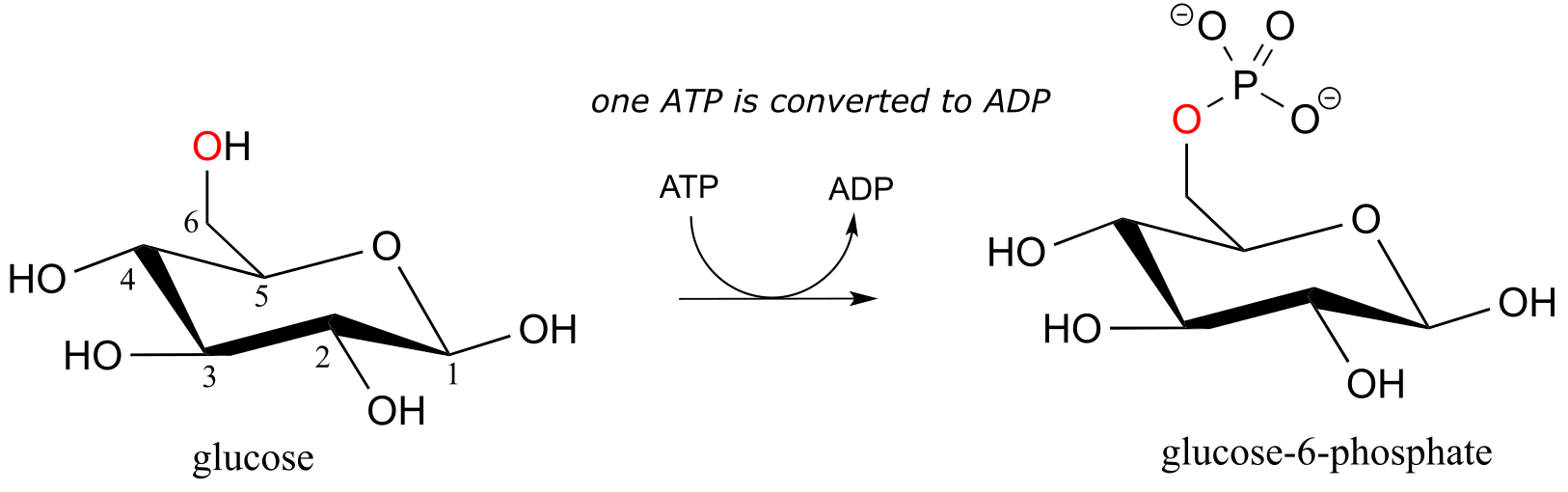
fig 26b
The biological activity of many proteins is regulated by protein kinases. In a protein kinase reaction, the side chain hydroxyl groups on serine, threonine, or tyrosine residues of certain proteins are phosphorylated by ATP:
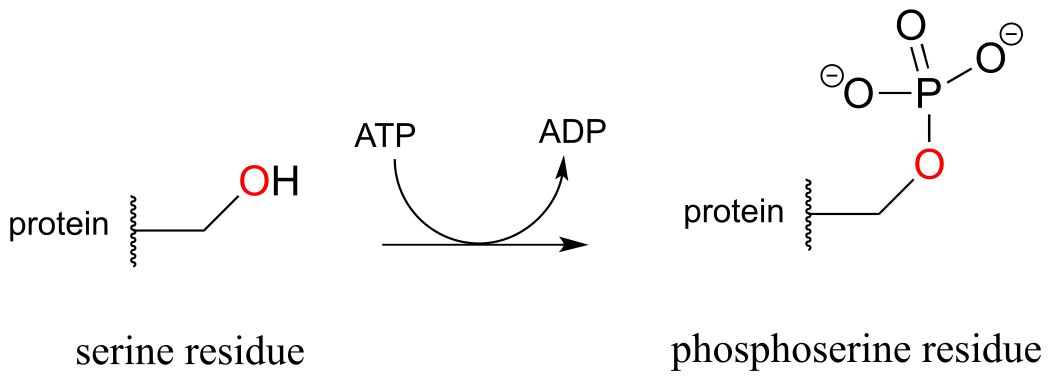
fig 27
The conversion of a neutral hydroxyl group to a charged phosphate represents a very dramatic change in the local architecture of the protein, potentially altering its folding pattern and ability to bind to small molecules or other proteins. A protein’s biological function can be ‘switched on’ by phosphorylation of a single residue, and switched off again by removal of the phosphate group. The latter reaction we will examine later in this chapter.
Exercise 9.9:
a) Draw a curved-arrow mechanism, using abbreviations as appropriate, for the serine kinase reaction.
b) Threonine kinase catalyzes the phosphorylation of the side chain hydroxyl group of threonine residues in proteins. Draw the structure, including the configuration of all stereocenters, of a phosphothreonine residue. Explain how you can predict the stereochemistry of the side chain.
Although stereochemical inversion in phosphate transfer is predicted by theory, the fact that phosphate groups are achiral made it impossible for a long time to verify the phenomenon directly. This was finally accomplished in the late 1970’s, when a group of researchers demonstrated phosphate inversion in kinase enzymes using chemically synthesized ATP in which three different isotopes of oxygen were incorporated into the γ−phosphate, thus creating a chiral phosphorus center. (Ann. Rev. Biochem. 1980 49, 877).
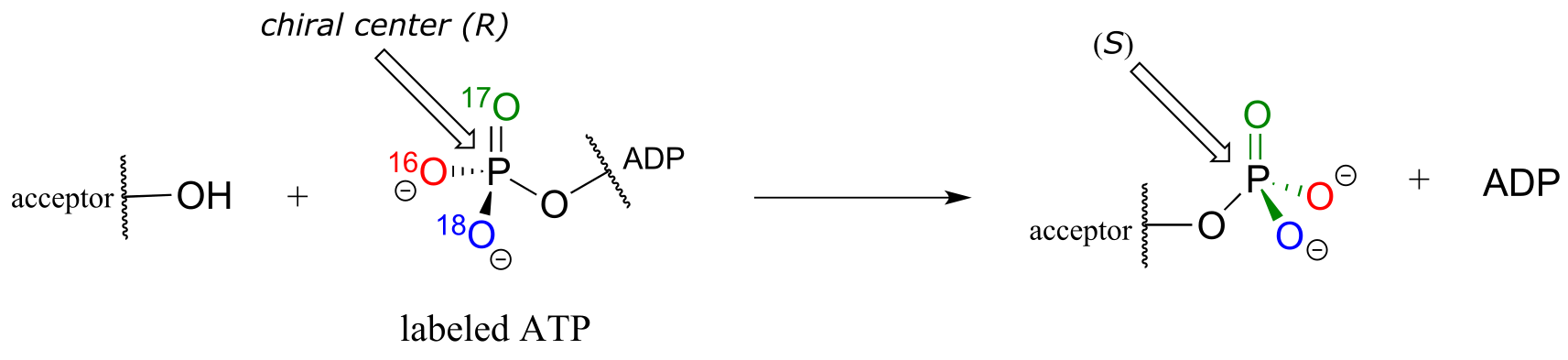
fig 27a
Alcohols can be converted into organic diphosphates in two different ways. A two-step process simply involves successive transfers of the γ-phosphate groups of two ATP donors, such as in these sequential steps in isoprenoid biosynthesis. (EC 2.7.1.36; EC 2.7.4.2). A compound called mevalonate is diphosphorylated in this way in the early phase of the biosynthetic pathway for cholesterol, steroid hormones, and other isoprenoid molecules.
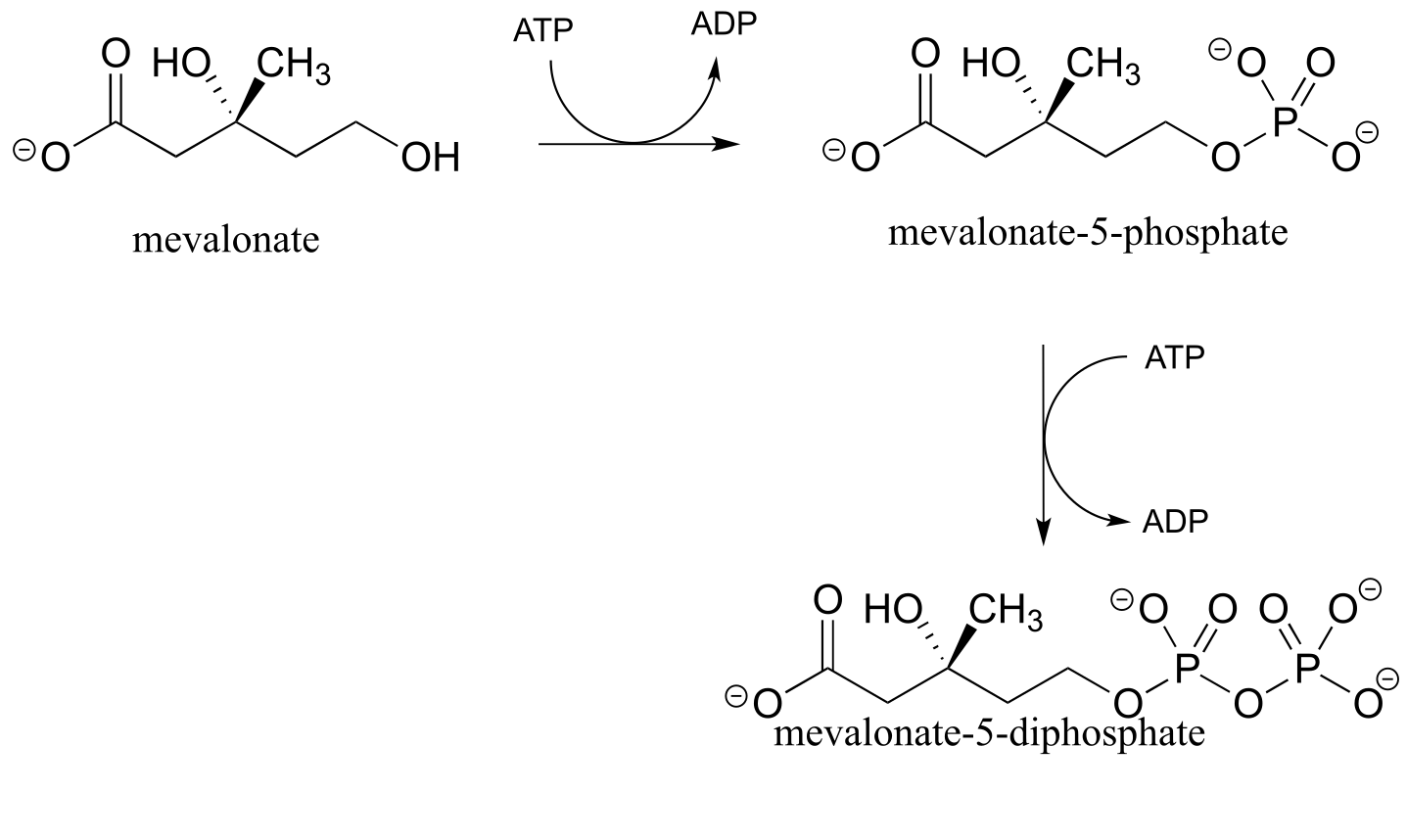
fig 28
The mechanism for the first phosphorylation step is analogous to that for an alcohol kinase reaction, which we have just seen. In the second phosphate transfer step, catalyzed by a separate enzyme, one of the phosphate oxygens on the organic monophosphate acts as a nucleophilic phosphate acceptor, attacking the γ-phosphate of a second ATP.
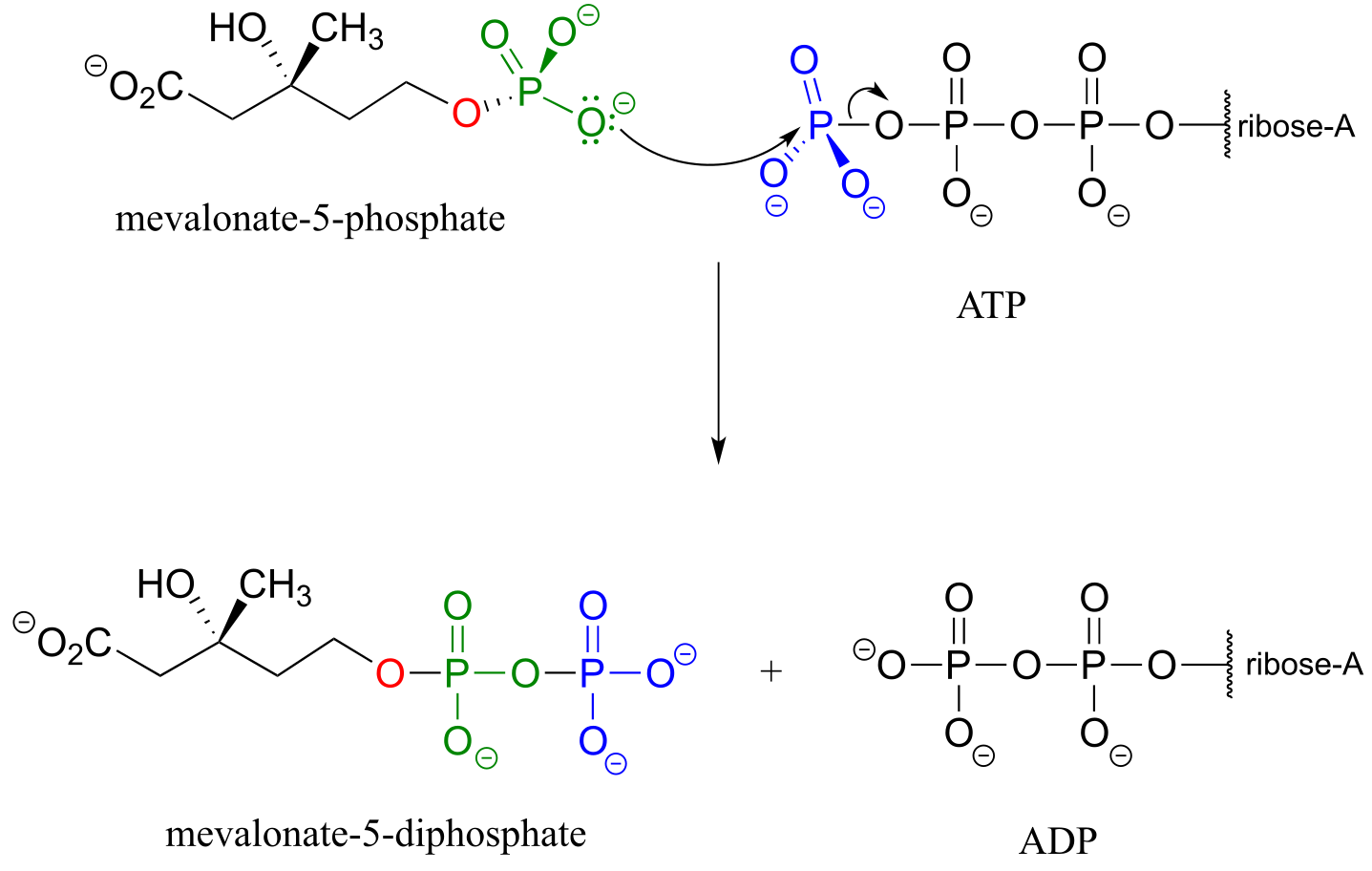
fig 29a
In some metabolic pathways, diphosphorylation occurs by a different mechanism from the one above. Instead of sequentially transferring two phosphates from two ATP donors, the alternate mechanism occurs in a single step: the nucleophilic acceptor molecule attacks the β-phosphate of ATP, rather than the γ-phosphate. After formation of the trigonal bipyramidal intermedia, it is AMP (not ADP) which is expelled, and what started out as the β and γ phosphates of ATP both remain with the acceptor.
In the biosynthesis of DNA and RNA nucleotides, one of the hydroxyl groups on ribose-5-phosphate is diphosphorylated (EC 2.7.6.1) in a one-step mechanism:
A one-step alcohol diphosphorylation reaction (PRPP synthase):
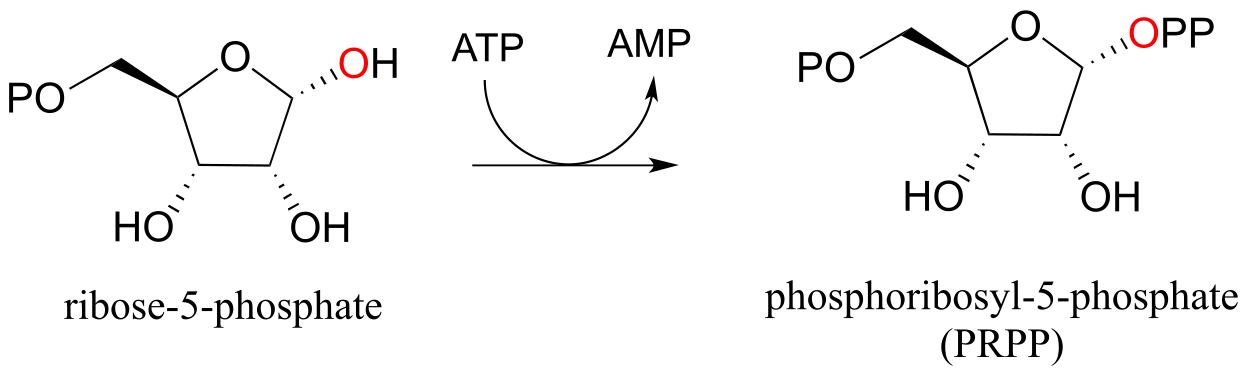
Mechanism:
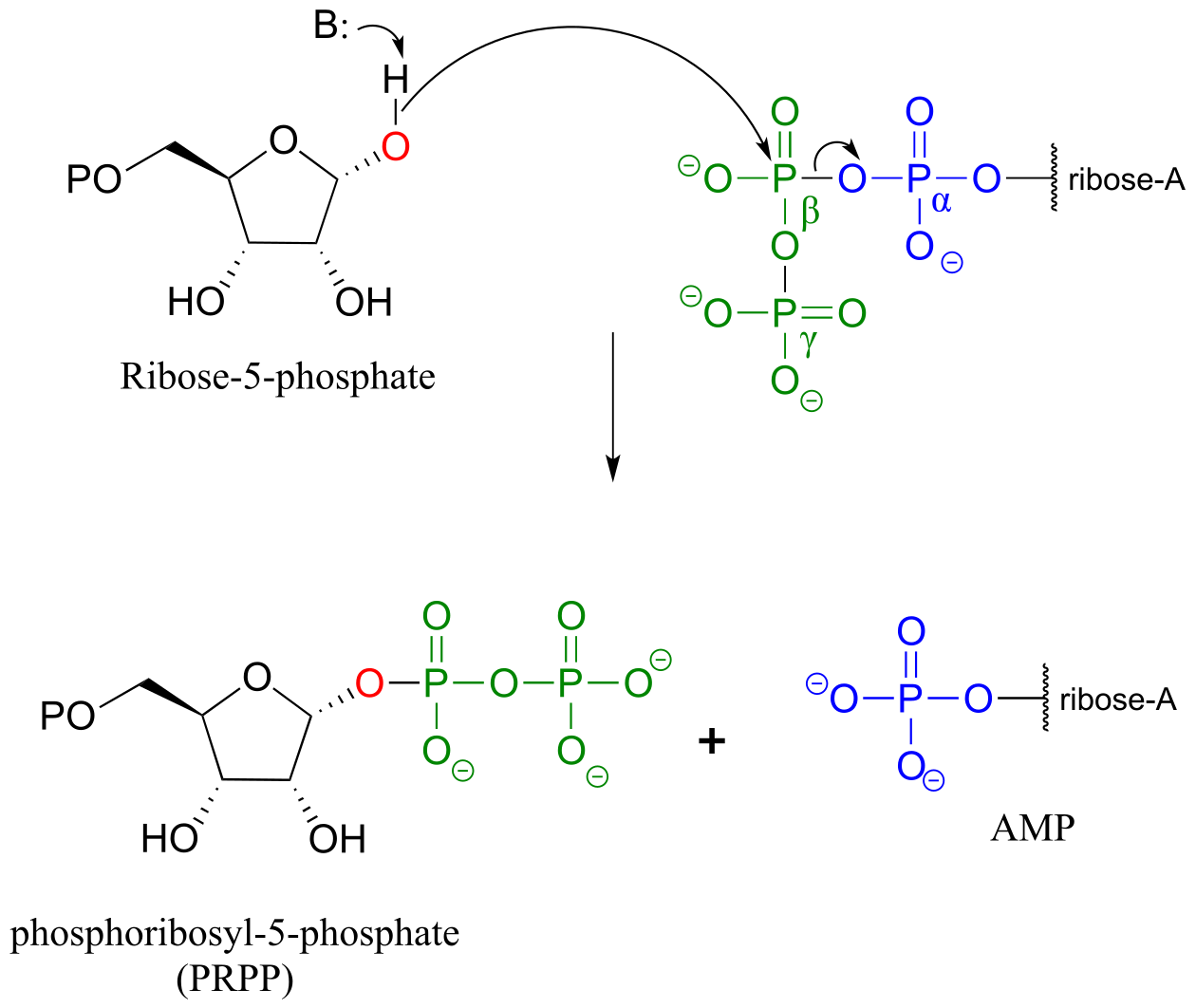
fig 31, 31b
The metabolic role of both of the diphosphorylation processes we just saw is to convert a hydroxyl group into a good leaving group (recall that hydroxide ions are strong bases and poor leaving groups, while phosphates/diphosphates, especially when stabilized in an enzyme active site, are weak bases and very good leaving groups). In nucleoside biosynthesis pathways, the diphosphate group of PRPP acts as a leaving group in the very next metabolic step, which is an SN1 reaction: we have already seen this reaction in section 8.7B).
9.5: Phosphorylation of carboxylates#
Thus far we have seen hydroxyl oxygens and phosphate oxygens acting as nucleophilic accepting groups in ATP-dependent phosphate transfer reactions. Carboxylate oxygens can also accept phosphate groups from ATP. This typically happens in two different ways. First, the carboxylate can attack the γ-phosphate of ATP to accept phosphate,
generates a species known as an ‘acyl phosphate’. An example is the first part of the reaction catalyzed by glutamine synthase (EC 6.3.1.2):
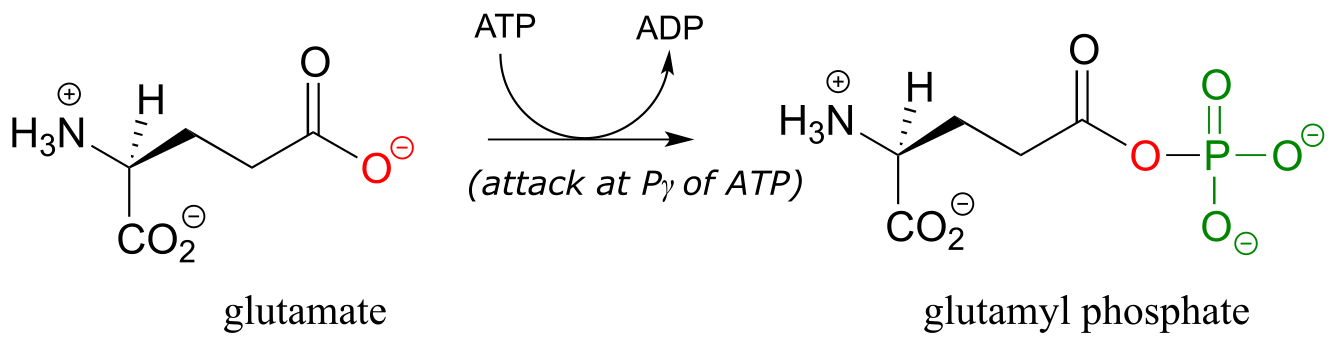
Alternatively, carboxylate groups are often converted into a species referred to as an ‘acyl-AMP’ . Here, the carboxylate oxygen attacks the α-phosphate of ATP leading to release of inorganic pyrophosphate. An example is the first part of the reaction catalyzed by the enzyme asparagine synthetase: (EC 6.3.5.4):
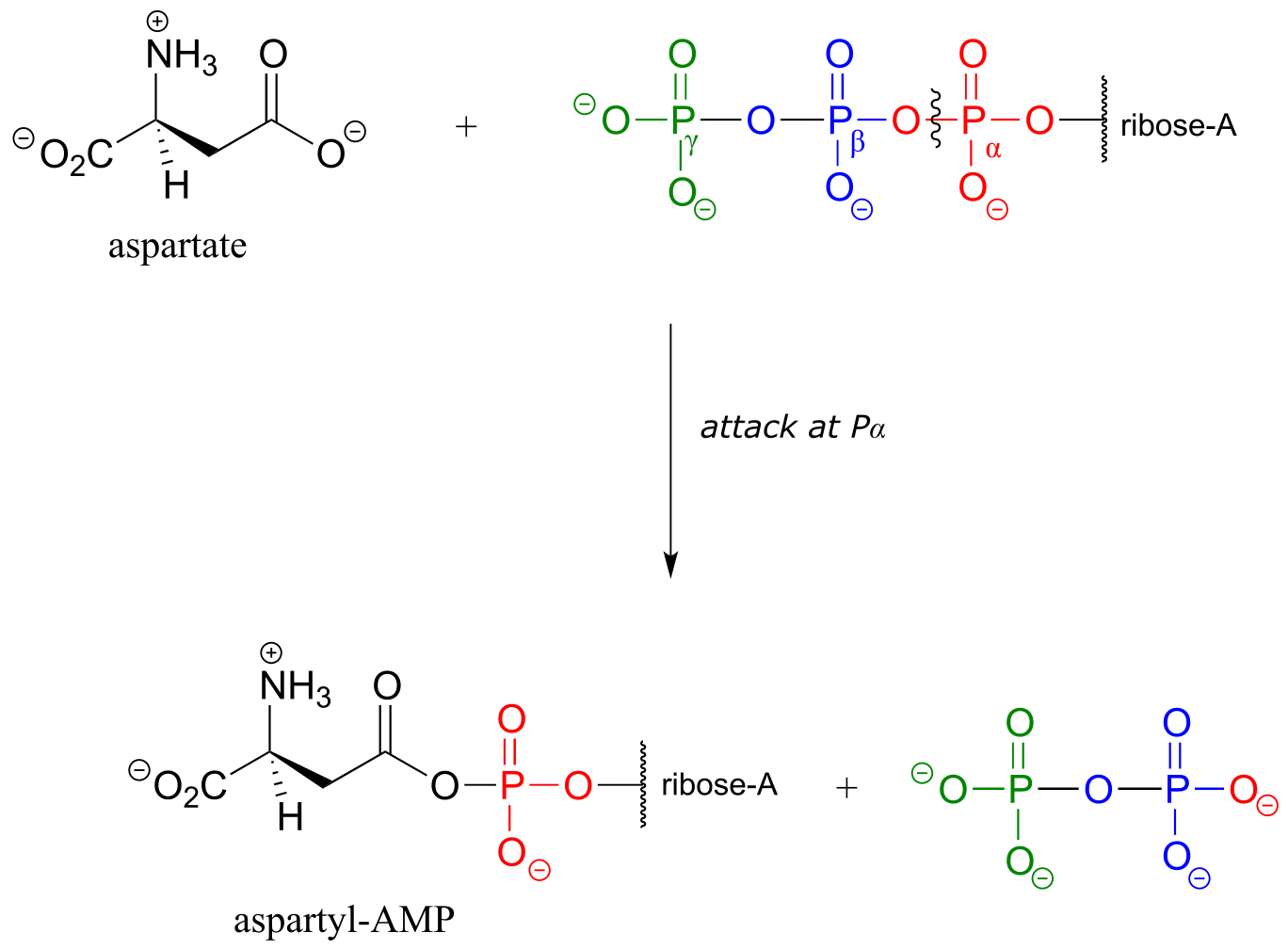
fig 35
Exercise 9.10: Draw a curved-arrow mechanism for the phosphate transfer reaction shown below (EC 2.7.2.3), which is from the glycolysis pathway. Note that ADP is on the reactant side and ATP is a product (the opposite of what we have seen so far). Hint: What functional group is the nucleophile? What functional group is the leaving group?
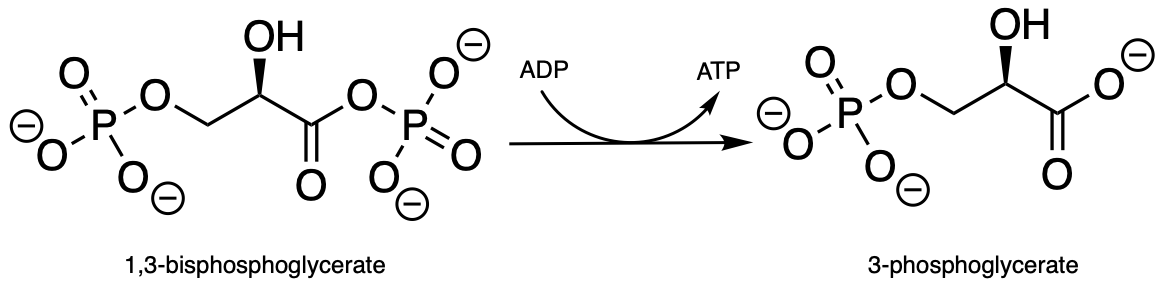
fig 36
9.6: Hydrolysis of organic phosphates#
While kinase enzymes catalyze the phosphorylation of organic compounds, enzymes called phosphatases catalyze dephosphorylation reactions. The reactions catalyzed by kinases and phosphatases are not the reverse of one another: kinases irreversibly transfer phosphate groups from ATP (or sometimes other nucleoside triphosphates) to various organic acceptor compounds, while phosphatases transfer phosphate groups from organic compounds to water: these are hydrolysis reactions. Kinase reactions involve an inherently ‘uphill’ step (phosphorylation of an alcohol, for example) being paid for with an inherently ‘downhill’ step (cleavage of an anhydride bond in ATP). Phosphatase reactions, on the other hand, are thermodynamically ‘downhill’, and while they require an enzyme to speed them up, they do not involve ‘spending’ energy currency the way kinase reactions do.
Phosphatase reaction:
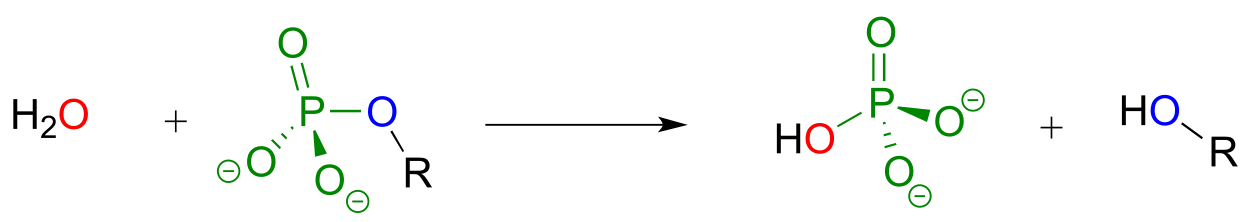
fig 37
There are two possible general mechanisms for a phosphatase reaction. Some enzymes catalyze direct hydrolysis reactions, in which the phosphate group is removed by direct attack of a water molecule at the phosphate center:
Phosphatase mechanism (direct hydrolysis):
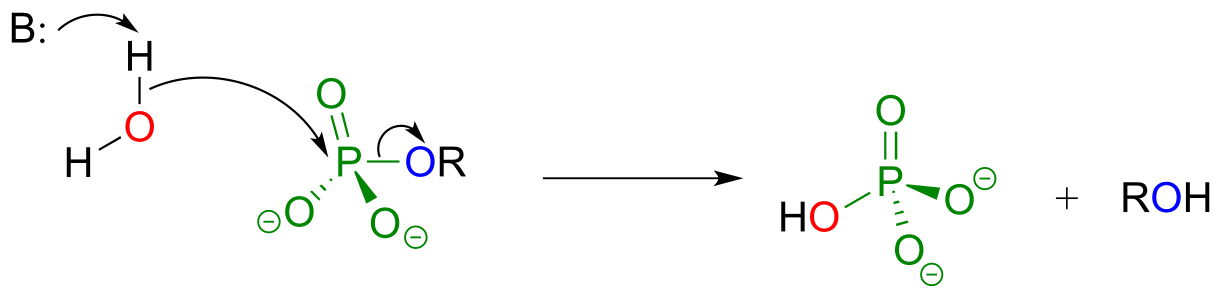
fig 37
One of the two phosphate groups on fructose 1,6-bisphosphate is hydrolyzed in such a way late in the gluconeogenesis pathway. (Biochemistry 2000, 39, 8565; EC 3.1.3.11)
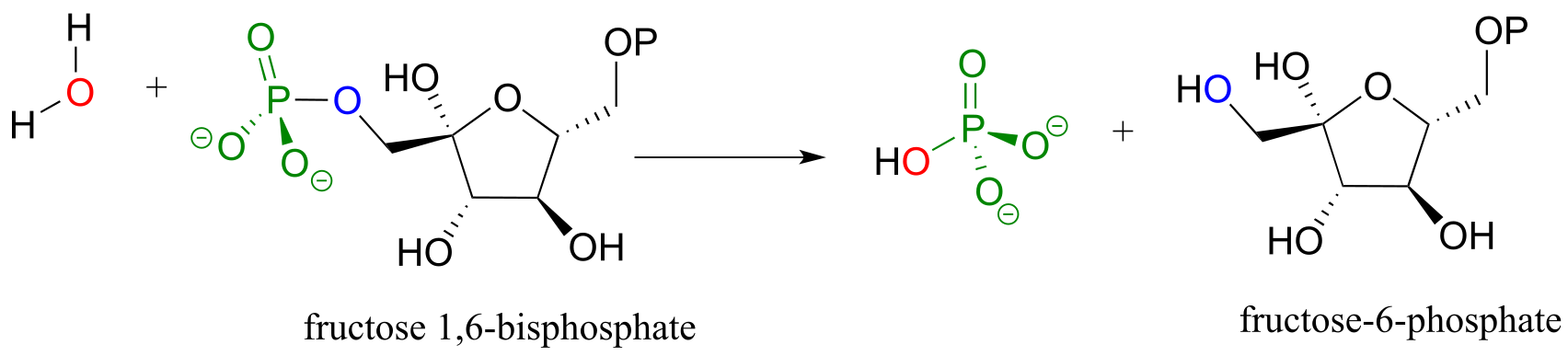
fig 37b
Many phosphatase reactions, however, operate by a slightly more complicated mechanism than what is shown above. In the first phase, a nucleophilic enzyme group (typically a cysteine, aspartate, glutamate, or histidine side chain, designated in the figure below as ‘X’) attacks the phosphate group. In the second phase, the phosphorylated residue is hydrolized. For example, protein tyrosine phosphatase catalyzes the dephosphorylation of phosphotyrosine residues in some proteins - this is the other half of the regulatory ‘on-off switch’ that we discussed earlier in the context of protein kinases. In the first step, the phosphate group is directly donated to a cysteine side chain in the phosphatase enzyme’s active site. In the second step, the phosphocysteine intermediate is cleaved by water to form inorganic phosphate and regenerate the free cysteine in the active site.
Indirect phosphatase reaction:
Step 1:
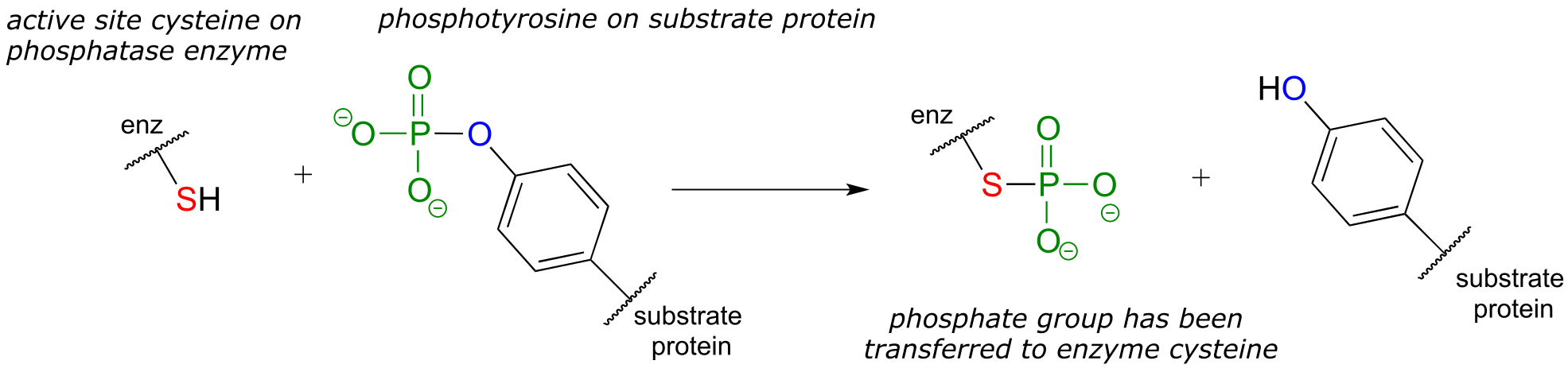
Step 2:
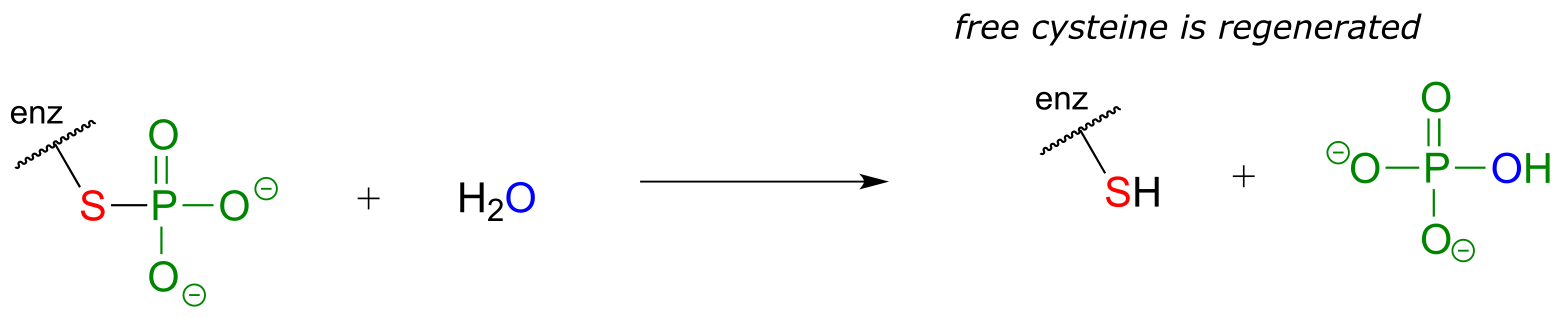
fig 38d
Notice that in the end, the phosphate group has still been transferred to a water molecule, albeit indirectly. How would you know, just by looking at the substrate and product of the protein tyrosine phosphatase reaction, that the phosphate is not transferred directly to a water molecule? Simply put, you wouldn’t know this information without the benefit of knowledge gained from biochemical experimentation.
Exercise 9.11: If you were to look just at the substrates and products of a phosphatase reaction without knowing anything about the mechanism, it is apparent that a nucleophilic substitution mechanism could theoretically account for the products formed. Draw out a hypothetical nucleophilic substitution mechanism for the hydrolysis of a phosphoserine residue and show how researchers, by running the reaction in H218O, (isotopically labeled water), could potentially distinguish between a nucleophilic substitution and phosphate group transfer mechanism by looking at where the 18O atom ends up in the products.
9.7: Phosphate diesters in DNA and RNA#
Phosphate diesters play an absolutely critical role in nature - they are the molecular ‘tape’ that connect the individual nucleotides in DNA and RNA via a sugar-phosphate backbone. Take note of the 1’ - 5’ carbon numbering shown below for the ribose sugar - these numbers will be used frequently in the coming discussion. The ‘prime’ symbol (’) is used to distinguish the ribose carbon numbers from the nucleotide base carbon numbers (which are not shown here).
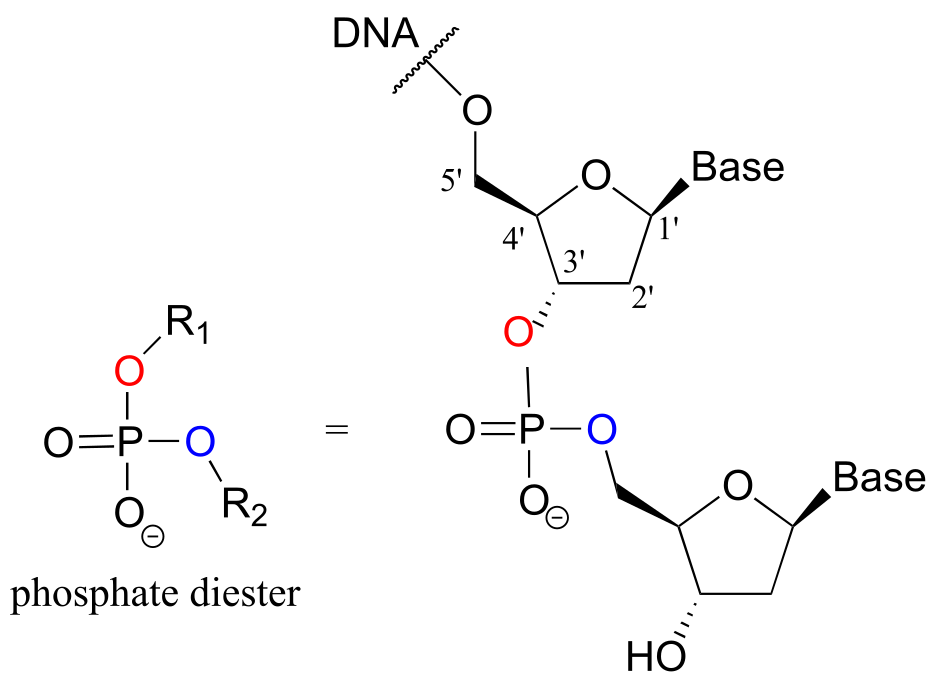
fig 5
The introduction to this chapter referenced a widely-read 1987 commentary in Science Magazine, in which F.H. Westheimer of Harvard University addressed the question of why phosphates were ‘chosen’ by nature for critical biochemical job of linking DNA (Science 1987, 235, 1173). He emphasizes how critical it is for the phosphate diester linkage in DNA to be stable in water – in other words, it must be resistant to spontaneous (nonenzymatic) hydrolysis. Even very infrequent occurrence of such an undesired hydrolysis event could be disastrous for an organism, given that an intact DNA strand is a long-term storage mechanism for genetic information.
Westheimer pointed out that the inherent stability of DNA is a due in large part to the negative charge on the non-bridging oxygen of the phosphate diester linker, which effectively repels nucleophilic water molecules and shields the electrophilic phosphorus atom from attack.
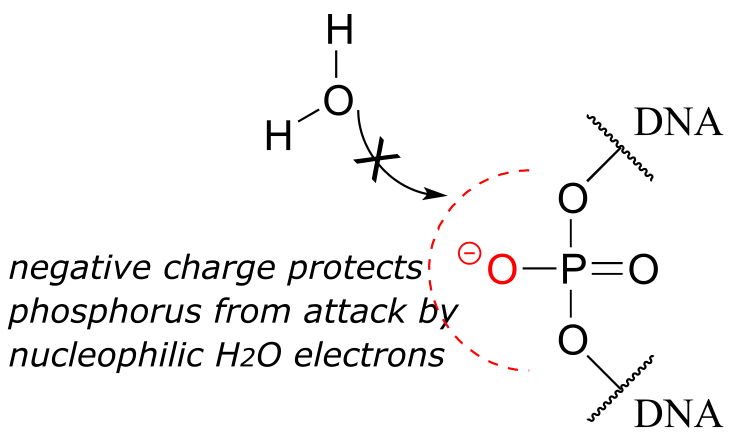
fig 40
While DNA is quite stable with regard to spontaneous hydrolysis, it of course can be degraded by specific enzymatic hydrolysis, where the phosphate electrophile is activated for attack through noncovalent interactions (eg. with Mg2+) in the active site. Enzymes that hydrolyze the phosphate diester bonds in DNA are called nucleases, and we will learn more about them in section 9.8.
Unlike DNA, RNA is quite vulnerable to spontaneous hydrolysis in aqueous solution. This does not present a physiological dilemma, because the function of RNA is to encode genetic information on a temporary rather than long-term basis. Why does hydrolysis occur so much more rapidly in RNA than in DNA? The answer has everything to do with the lowered entropic barrier to the reaction (you might want to quickly review the concept of entropy at this point). RNA nucleotides, unlike the deoxynucleotides of DNA, have a hydroxyl group at the neighboring 2’ carbon. The 2’ hydroxyl group is right next to the electrophilic phosphorus atom, poised in a good position to make a nucleophilic attack, breaking the RNA chain and forming a cyclic phosphate diester intermediate (see figure below).

fig 41
Researchers working with RNA have to be careful to store their samples at very cold temperatures, preferably freeze-dried or precipitated in ethanol, to avoid hydrolysis. The problem of RNA decomposition is compounded by the fact that RNAase enzymes, which catalyze RNA hydrolysis, are present on the surface of human skin and are very stable, long-lived, and difficult to destroy.
In contrast, DNA samples can be safely stored in aqueous buffer in a refrigerator, or in a freezer for longer-term storage.
9.8: The organic chemistry of genetic engineering#
Many enzymes that catalyze reactions involving the phosphate diester bonds of DNA have been harnessed for use in genetic engineering - techniques in which we copy, snip, and splice DNA in order to create custom versions of genes. The tools of genetic engineering have become indispensable and commonplace in the past decade, and most researchers working on the biological side of chemistry use them extensively. The days of painstakingly purifying an enzyme from bacterial cultures or ground-up cow livers are pretty much gone. Now scientists clone the gene that encodes the enzyme, make any desired changes (by site-directed mutagenesis, for example), and use a host such as E. coli or yeast to produce ‘recombinant’ enzyme from the cloned gene. You will learn the details of many of these procedures in a biochemistry or molecular biology course. What we will focus on now is applying what we have learned about phosphate group transfer reactions so that we can recognize some of the organic chemistry that is happening in a cloning experiment.
The first thing you have to do in a gene cloning procedure is to copy a DNA strand. This is accomplished by an enzyme called DNA polymerase (EC 2.7.7.7), which uses a single strand of DNA as a template to synthesize a second, complementary strand (the full picture of this complex process is well beyond the scope of this book, but recall that we talked about the discovery of thermostable DNA polymerase in the introduction to chapter 6).
You may have learned in a biology class that DNA is synthesized in the 3’ to 5’ direction. Notice below that the 3’ hydroxyl group on the end of the growing DNA strand attacks the α-phosphate of a 2’-deoxynucleoside triphosphate (dNTP), expelling inorganic pyrophosphate.
DNA polymerase reaction:
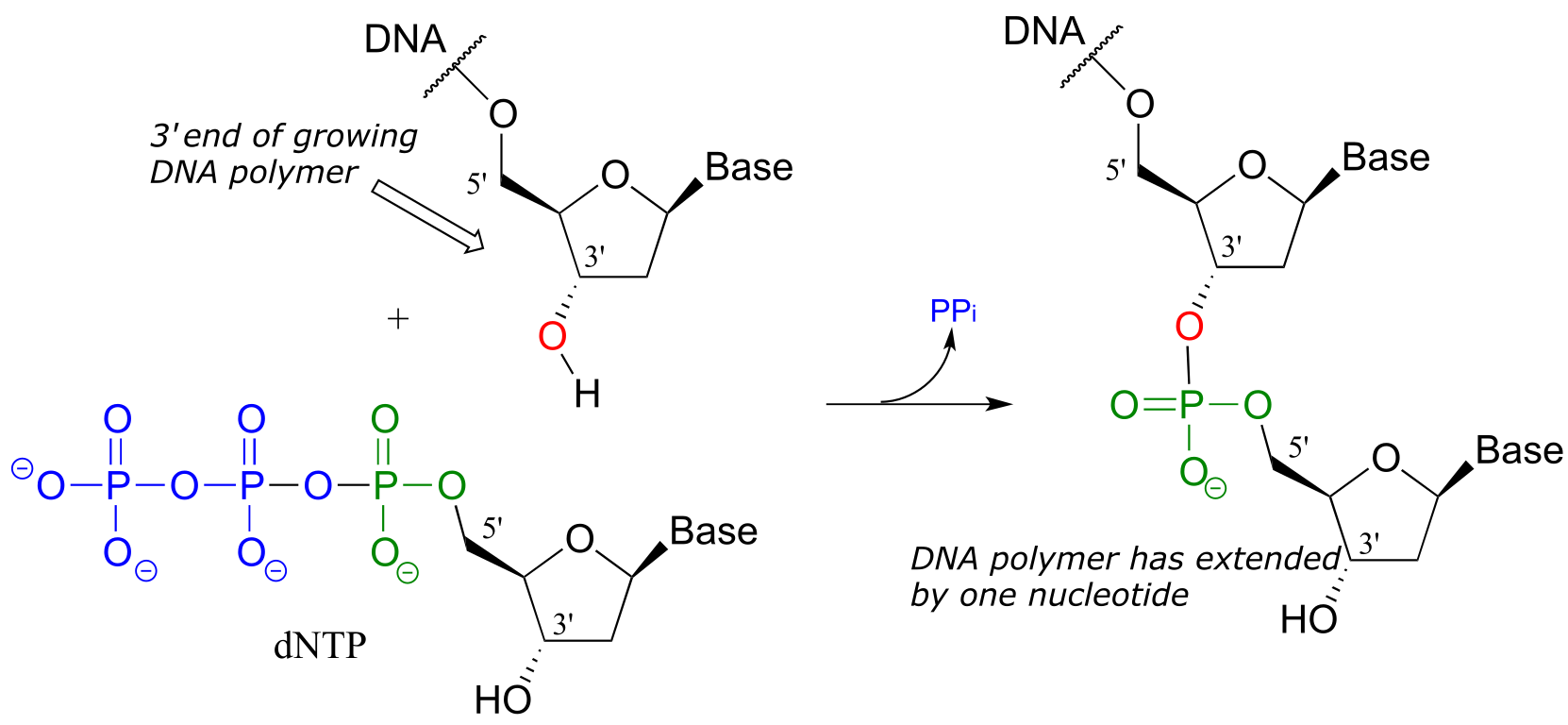
fig 42
Scientists are able to cut DNA using ‘molecular scissor’ enzymes called restriction endonucleases that cleave double-stranded DNA by hydrolysis at specific base sequences.
DNA hydrolysis by restriction endonucleases:
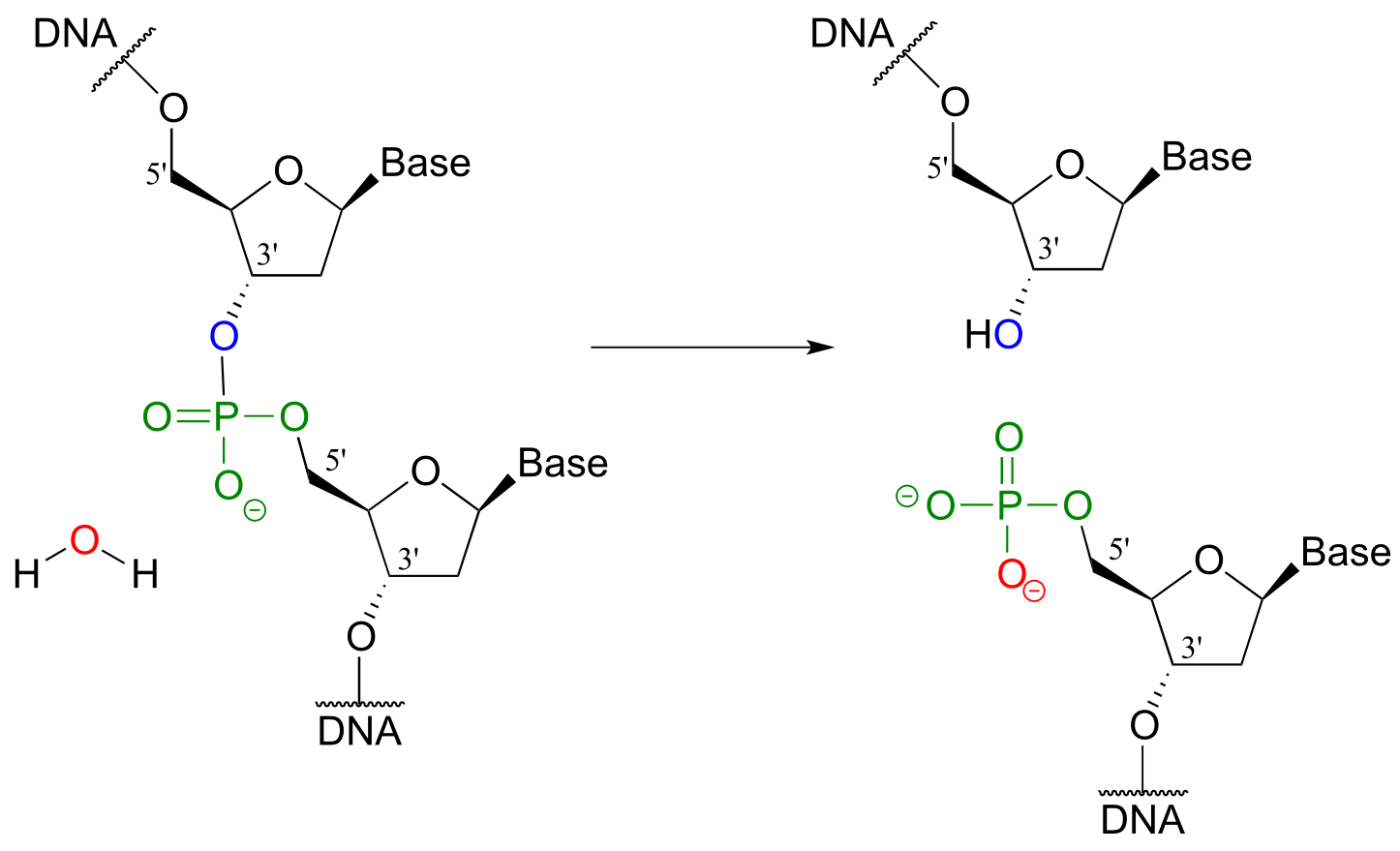
fig 43
Notice that the result of this hydrolytic cleavage reaction is one segment of DNA with a hydroxy group at the 3’ position, and a second segment with a phosphate group at the 5’ position.
A commonly used restriction endonuclease called ‘BamHI’ cleaves double-stranded DNA specifically at the following 6-base sequence:
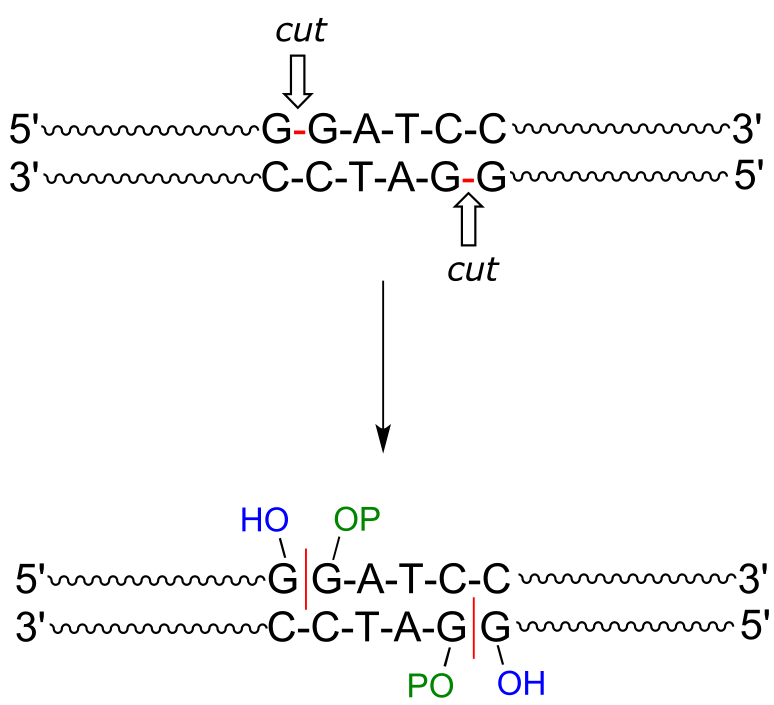
fig 43
Notice that a ‘staggered’ cut is made: this is a common (and useful) property of many endonucleases, although some make ‘blunt-ended’ cuts.
While an endonuclease cleaves a phosphodiester linkage in a DNA strand, DNA ligase (EC 6.5.1.1) accomplishes the reverse process: it catalyzes the formation of a new 3’-5’ link between two strands:
DNA ligase reaction:
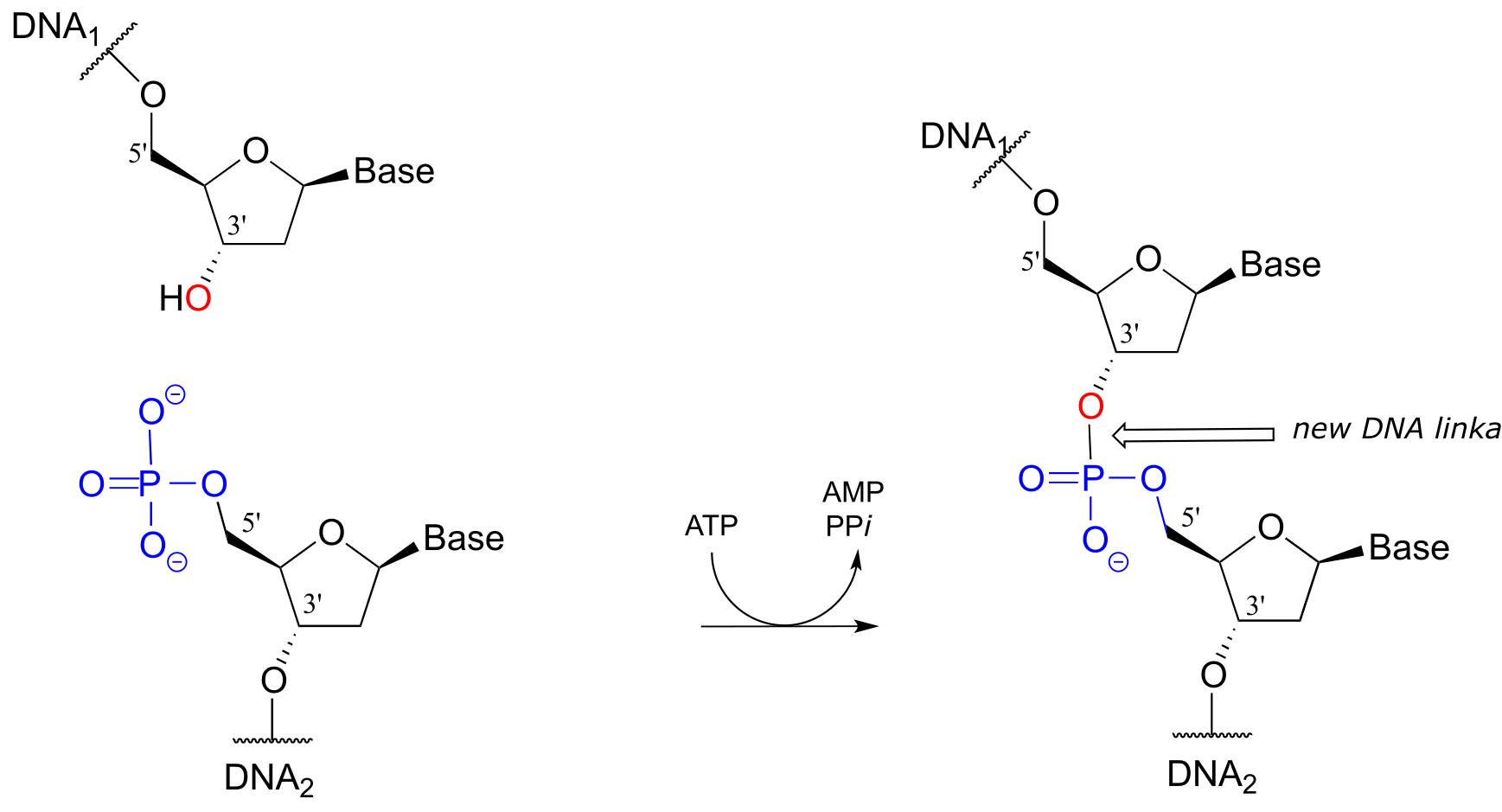
fig 45a
Note that there is initially no leaving group on the 5’ phosphate of DNA2, which makes a direct phosphate transfer reaction impossible. The strategy employed by the DNA ligase enzyme is to first activate the 5’ phosphate of DNA2 using ATP (phase 1 below), then the ligation reaction can proceed (phase 2)
DNA ligation
Phase 1: activation
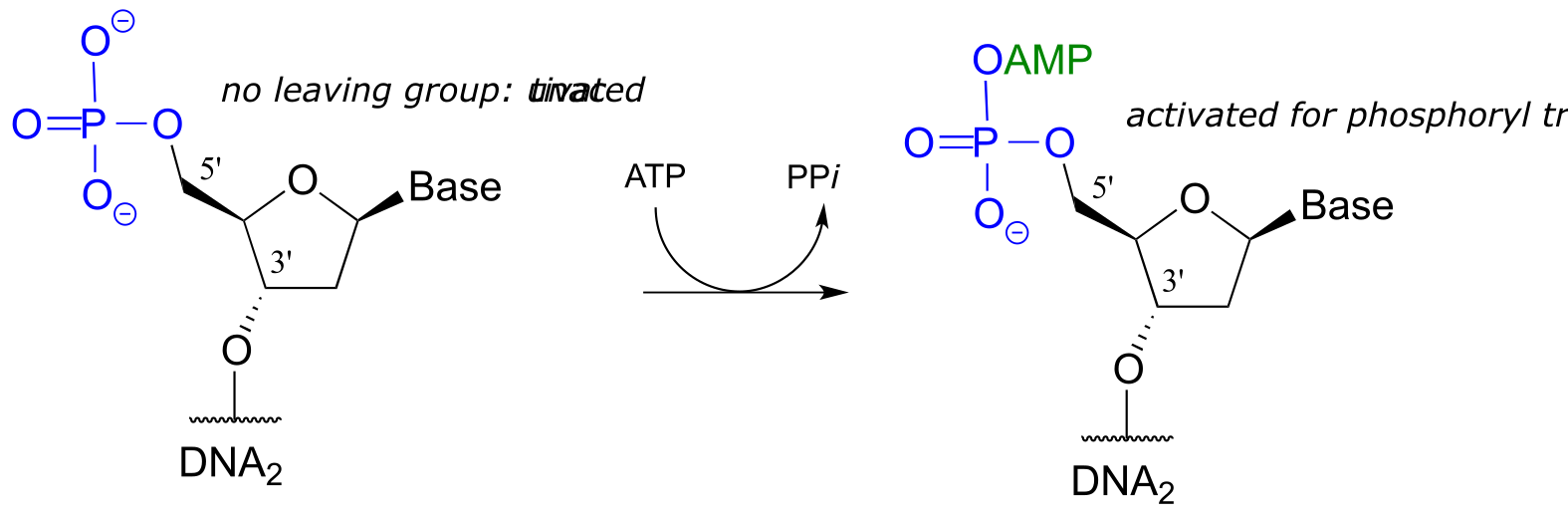
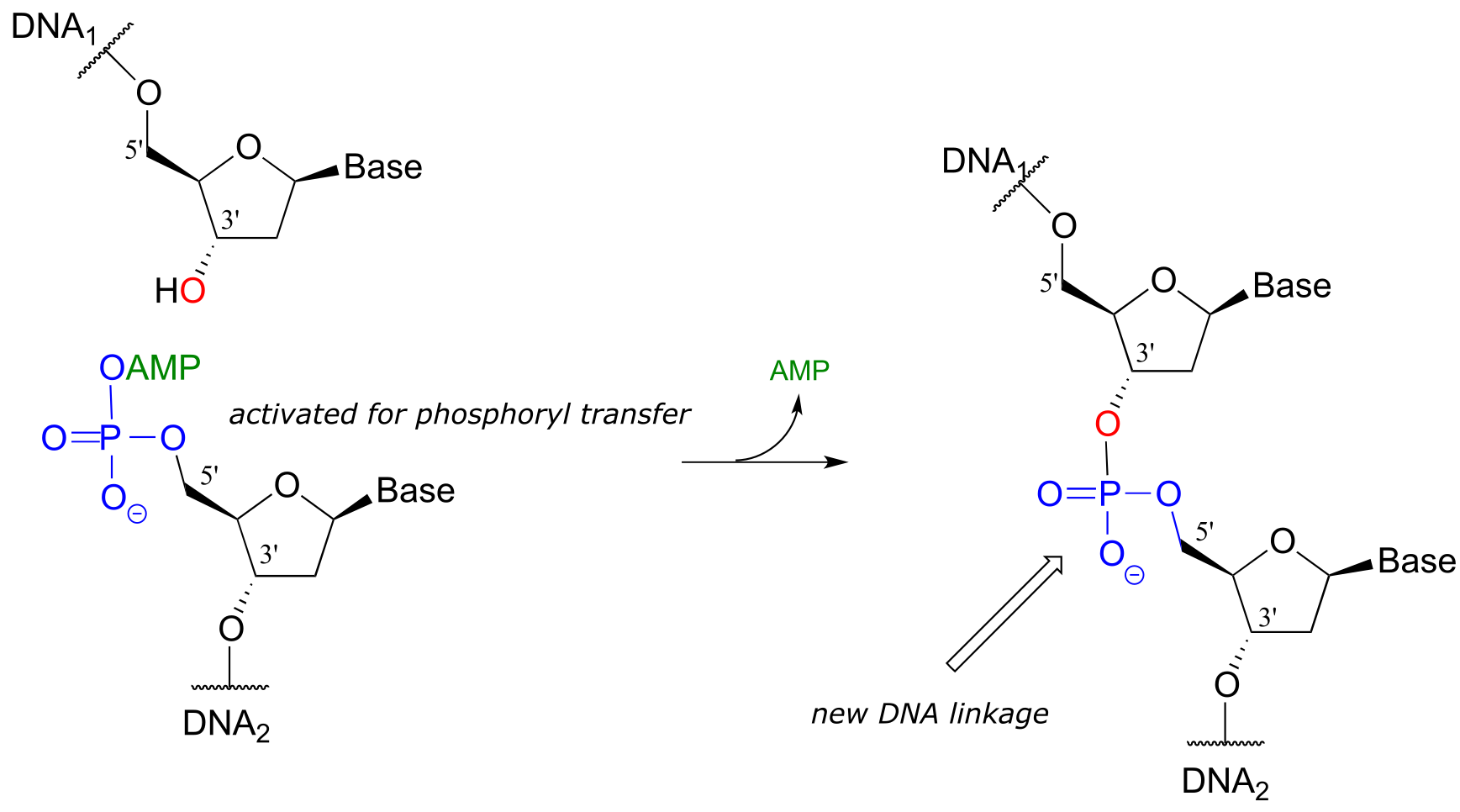
Phase 2: ligation:
One more enzymatic tool in the genetic engineering arsenal bears mention. In some cloning procedures, a researcher may want to prevent unwanted ligation of DNA. This can be accomplished by using the enzyme alkaline phosphatase (EC 3.1.3.1), which catalyzes the dephosphorylation of many different organic phosphates, including 5’-phosphorylated DNA (recall that we discussed phosphatases in section 9.6).
Alkaline phosphatase reaction:

fig 46
With the phosphate group removed, ligation is impossible - there is no way to make a new phosphodiester bond without a 5’ phosphate group!
**
**
9.9: NMR of phosphorylated compounds#
Because so many biological molecules contain phosphoryl groups, it is worthwhile to look at how scientists use NMR to determine the structure of these molecules. Recall from section 5.1 that 31P, the most abundant isotope of phosphorus, is NMR active: it can be directly observed by 31P-NMR, and indirectly observed in 1H-NMR and 13C-NMR through its spin-coupling interactions with neighboring protons and carbons, respectively.
Consider the case of isopentenyl diphosphate, the building block molecule used by cells to make ‘isoprenoid’ compounds such as cholesterol (in many animals), or β-carotene (in some plants). NMR spectra of this molecule were taken in a D2O solvent, buffered with ND4OD (the deuterium equivalent of aqueous ammonium hydroxide, NH4OH) (J. Org. Chem. 1986, 51, 4768). In our discussion, carbon atoms are specified with numbers, protons with lower case letters, and phosphorus atoms with upper case letters.
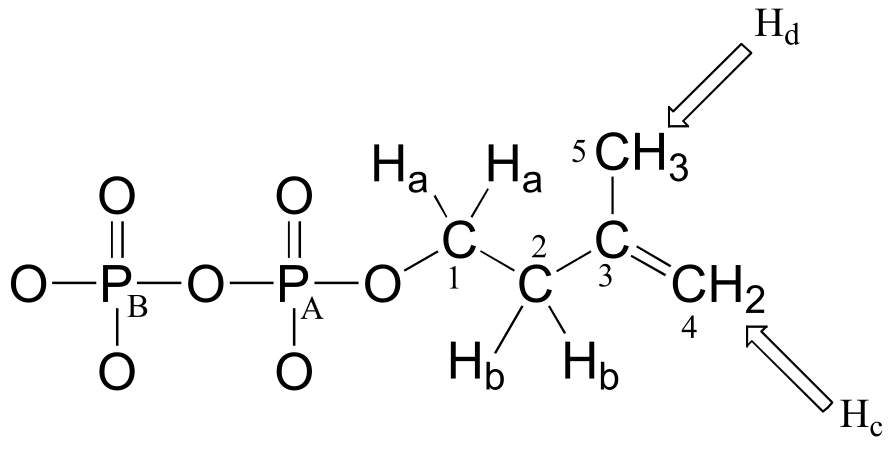
First, let’s look at the proton spectrum:
1H-NMR
Ha: 4.05 ppm (td); 3JHa-Hb = 6.6 Hz; 3JHa-PA = 3.3 Hz.
Hb: 2.39 ppm (t) 3JHa-Hb = 6.6 Hz
Hc: 4.86 ppm (s)
Hd: 1.77 ppm (s)
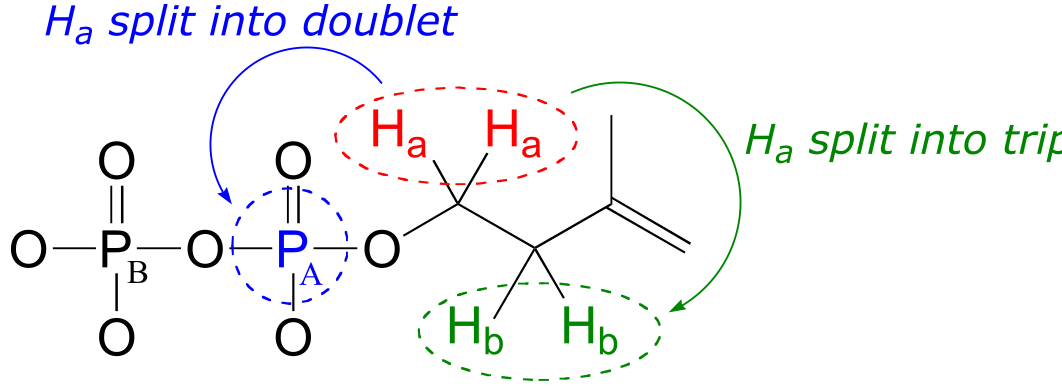
The signals for Hb, Hc, and Hd look like we would expect from our discussion in chapter 5, with the exception of Hc which you will be invited to discuss in the exercise below.
Why, though, is the signal for Ha split into a triplet of doublets (td)? First of all, as, expected, the two neighboring Hb protons split the Ha signal into a triplet, with 3JH-H = 6.6 Hz. Then, the signal is further split into doublets (3JH-P = 3.3 Hz) by PA, the closer of the two phosphorus atoms. A phosphorus atom will spin-couple with protons up to three bonds away.
Exercise 9.12: The signal for the two ‘Hc’ protons in isopentenyl diphosphate is reported above as a singlet integrating to 2H. Are these two protons really chemically equivalent, and, according to what you know about proton NMR, should this signal really be a singlet? If not, what kind of signal(s) would you expect to see? Explain any discrepancies between what you would expect to see and the actual reported data.
Now, let’s look at the 13C spectrum of IPP:
13C-NMR (proton-decoupled)
C1: 40.7 ppm (d); 2JC1-PA = 7.2 Hz
C2: 67.0 ppm (d); 3JC2-PA = 4.0 Hz
C3: 147.4 ppm
C4: 114.6 ppm
C5: 24.5 ppm
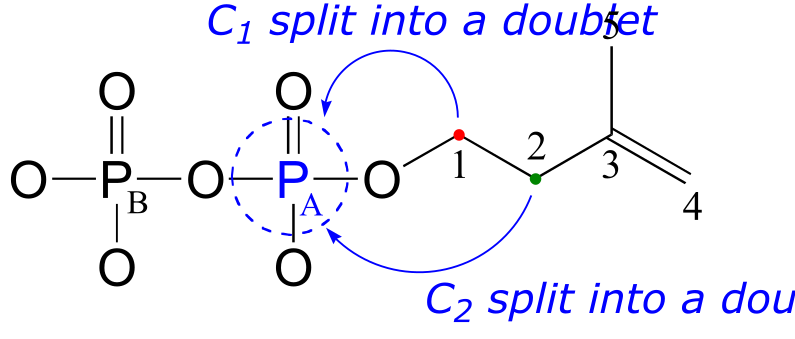
Notice that the signals for both C1 and C2 are split into doublets by the magnetic field of PA. Phosphorus atoms will spin-couple with 13C nuclei up to three bonds away. Notice also that the 2-bond coupling between C1 and PA is larger than the 3-bond coupling between C2 and PA (7.2 Hz vs. 4.0 Hz). Finally, notice that we do not observe 4-bond C-P coupling: C3 is not spin-coupled to PA, and PB is not coupled to any of the 13C or 1H nuclei on the molecule.
Remember that when processing a typical 13C-NMR spectrum, we electronically ‘turn off’ spin coupling between carbons and neighboring protons in order to simplify the spectrum (this is referred to as ‘proton decoupling’). Proton decoupling does not turn off C-P spin coupling.
Because 31P is NMR-active, we can also, with an NMR spectrophotometer equipped with a phosphorus probe, directly observe the phosphorus NMR signals, just as we can directly observe the signals from protons and 13C nuclei. On an NMR instrument where protons resonate at 300 MHz and 13C nuclei resonate at 75 MHz, phosphorus resonates at 32 MHz. In 31P-NMR experiments, the reference standard used to determine the 0 ppm point is usually phosphoric acid (tetramethylsilane, the standard 0 ppm point for 1H- and 13C-NMR, doesn’t have a phosphorus atom!). The 31P-NMR spectrum of isopentenyl diphosphate has, as expected, two peaks, each of which is upfield of the phosphoric acid standard (negative chemical shifts!) and split into a doublet (2JP-P = 20 Hz) due to 2-bond coupling between the two phosphorus nuclei.

fig 13
PA: (-)11.03 ppm (d, 2JP-P = 20 Hz)
PB: (-)7.23 ppm (d, 2JP-P = 20 Hz)
Notice that although the C1 and C2 signals were split by PA in our 13C-NMR spectrum, in the 31P-NMR spectrum the converse is not true: the PA signal is not split by C1 or C2. Both of these carbons are NMR-inactive 12C isotope in 99 out of 100 molecules. In addition, P-H splitting is not observed in this 31P spectrum, because proton decoupling is in effect.
Key concepts to review#
All of the reactions detailed in this chapter involved the transfer of a phosphate group - usually a phosphate, diphosphate, or AMP group - from one molecule (the donor) to another (the acceptor). Your learning goal for this chapter is to recognize and understand what is happening in these phosphate group transfer reactions, and to gain a basic understanding of the chemistry of phosphate and other phosphate groups.
Be sure that you can identify and provide examples of the following terms:
inorganic phosphate |
organic triphosphate |
|---|---|
inorganic phosphate |
phosphate (mono)ester |
inorganic pyrophosphate |
phosphate diester |
organic (mono)phosphate |
phosphate anhydride |
organic diphosphate |
bridging/non-bridging oxygen |
Also, make sure that you can recognize and use appropriately the various abbreviations introduced in this chapter:
Pi PPi R-OP
R-OPP R-OAMP ATP
ADP AMP
… in addition to the various structural abbreviations for adenosine mono-, di-, and triphosphate.
You should know the approximate pKa values for phosphoric acid and an organic monophosphate, and be able to state the approximate net charge (to the nearest 0.5 charge unit) of these species in buffers of different pH levels.
You should be able to describe, in words and pictures, the tetrahedral sp3d bonding picture for the phosphorus atom of a phosphate group. Even though the geometry is not always shown in every drawing, always keep in mind that the phosphate group is tetrahedral.
You should be able to draw resonance contributors for different phosphate groups, identify major versus minor contributors, and explain why some are major and some are minor. Remember - charges are shared between non-bridging oxygens, even if they are not drawn that way!
Absolutely critical to your success with this chapter is being able to picture and illustrate the mechanistic pattern which we refer to as a phosphate group transfer.
Though not usually included in reaction illustrations, always remember that charge-charge interactions with magnesium ions and hydrogen bonds to active site amino acids both serve to increase the electrophilicity of a phosphorus atom in donor compounds such as ATP.
You should understand the distinctions between the three mechanistic models for phosphate transfer reactions - concerted, addition-elimination, and elimination-addition - and know that the concerted model probably most closely describes biochemical reactions.
You should be able to identify the apical and equatorial positions in the pentavalent transition state of a phosphate transfer reaction, and recognize that the reaction results in inversion at the phosphorus center.
You should be able to identify the α, β, and γ phosphate groups of ATP, as well as the ribose and adenosine parts of the molecule.
You should be able to explain how ATP acts as a phosphate group donor, and why such reactions are thermodynamically favorable.
You should be able to draw a curved-arrow mechanism for reactions in which ATP acts as a phosphate group donor in the phosphorylation and/or diphosphorylation of an alcohol. You should be able to predict the result of nucleophilic attack at the α, β, or γ phosphates of ATP.
In general, you should be able to propose a likely mechanism for any phosphate transfer reaction, given the starting compounds and products.
Given information about the existence of a covalently linked enzyme-substrate complex in an enzyme mechanism, you should be able to propose a likely mechanism that accounts for such an intermediate. For example, after being told that the phosphotyrosine phosphatase reaction involves a phosphocysteine intermediate, you should be able to propose a mechanism.
Problems#
In all of the problems that follow, feel free to use appropriate abbreviations when drawing structures. However, always be sure not to abbreviate regions of a structure which are directly involved in bond-breaking or bond-forming events.
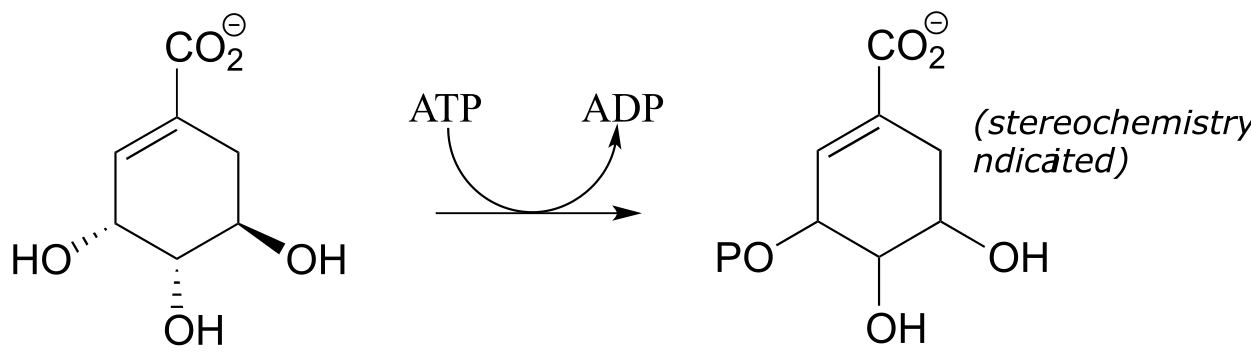
P9.1: Draw a likely mechanism for reaction catalyzed by shikimate kinase (EC 2.7.1.71) in the aromatic amino acid biosynthesis pathway). Stereochemistry of the product is not indicated in the figure below - in your mechanism, show the stereochemistry of the product, and explain how you are able to predict it from your knowledge of kinase reactions.
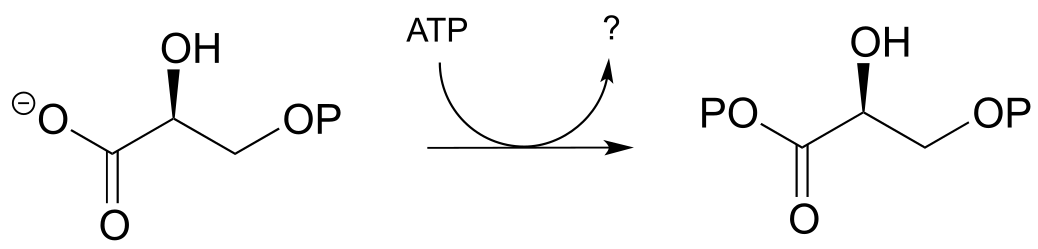
P9.2: Draw a likely mechanism for the following reaction (EC 2.7.2.3) in the gluconeogenesis pathway, and predict what compound is indicated by the question mark.
P9.3:
a) Draw a likely mechanism for the following reaction (EC 6.3.4.2) from ribonucleotide biosynthesis. Hint: what is the nucleophilic group? How could the enzyme increase it’s nucleophilicity?
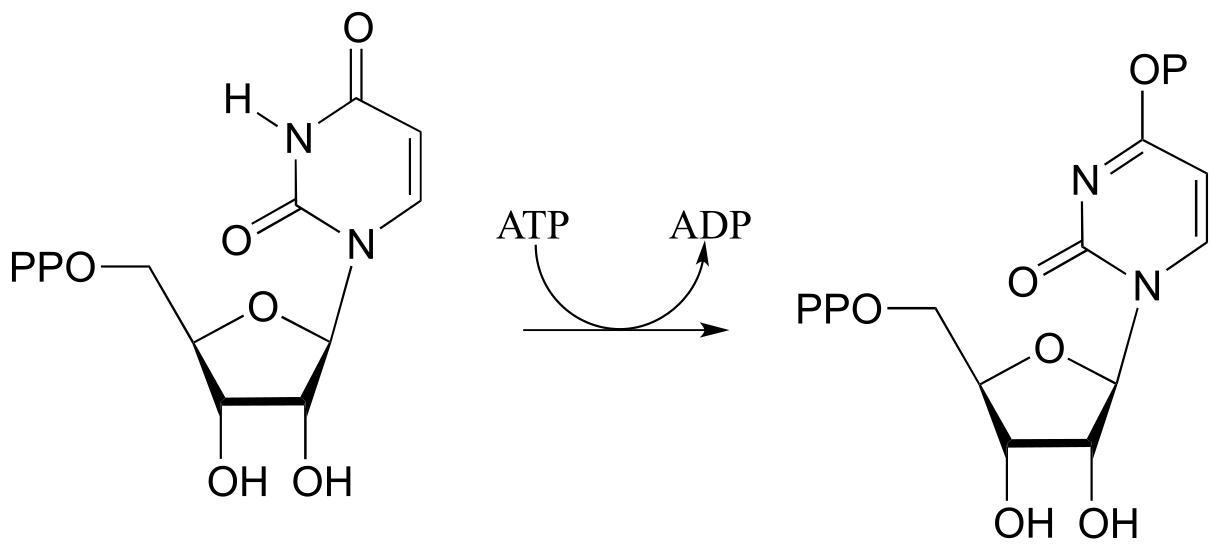
b) Draw a mechanism for the following reaction, also from ribonucleotide biosynthesis (EC 6.3.3.1):
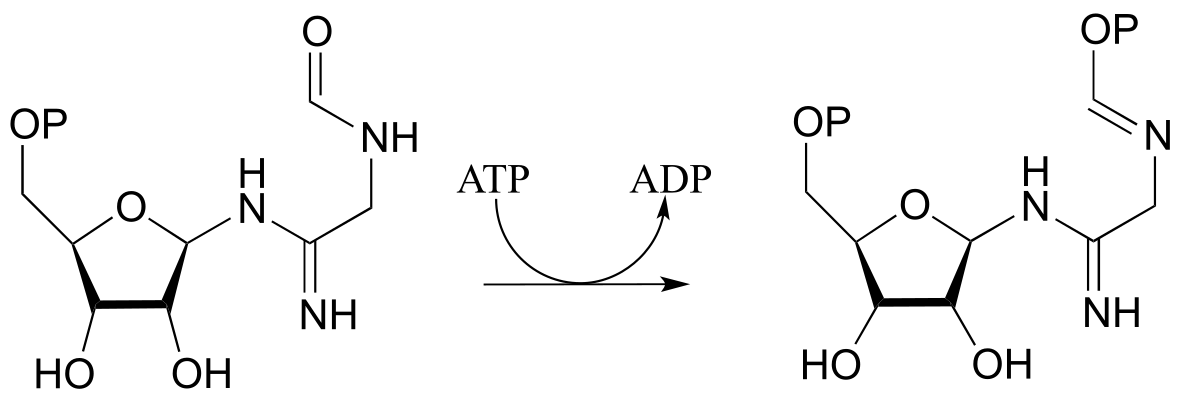
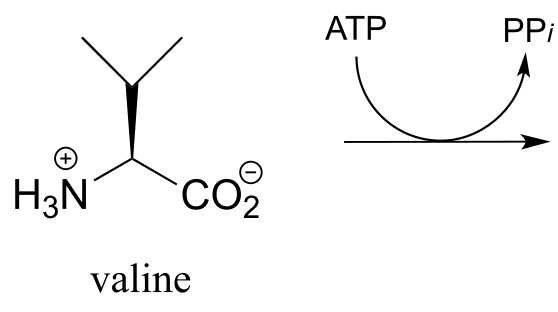
P9.4:
a) The carboxylate group on the amino acid valine is activated in an early step in the biosynthesis of the antibiotic penicillin. Predict the product of this reaction, and draw the likely intermediate of the phosphate group transfer reaction.
P9.5: The reaction below is an early step in the synthesis of tyvelose, a sugar found on the surface of some pathogenic bacteria. Notice that CTP plays the role of the phosphate group donor in this case, rather than ATP.
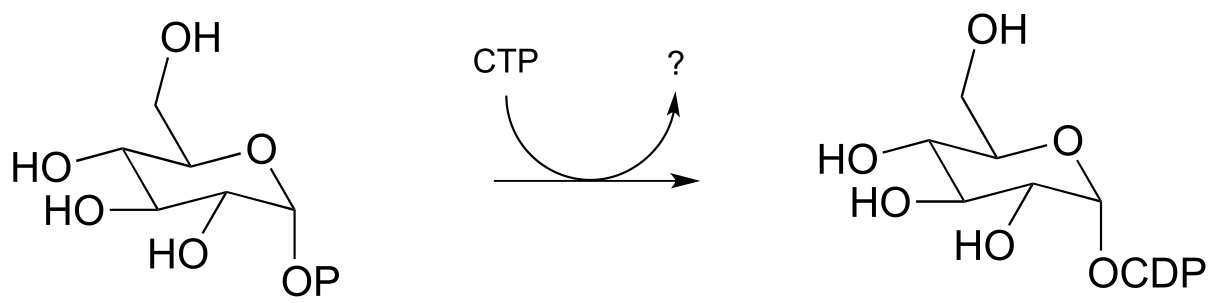
Draw a mechanism for the reaction, and indicate the second product that is released by the enzyme. (J. Biol. Chem. 2005, 280, 10774)
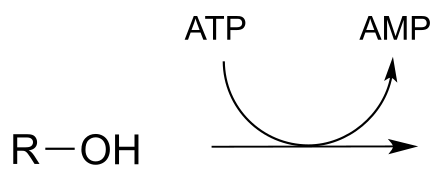
P9.6: Draw the likely product of the following hypothetical phosphate group transfer reactions. Specify which phosphate group of ATP is the electrophile in each case.
a)
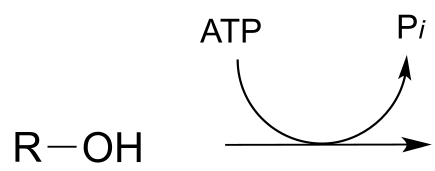
b)
P9.7: The figure below illustrates an experiment in which a reaction catalyzed by an E. coli enzyme was run in isotopically labeled water.
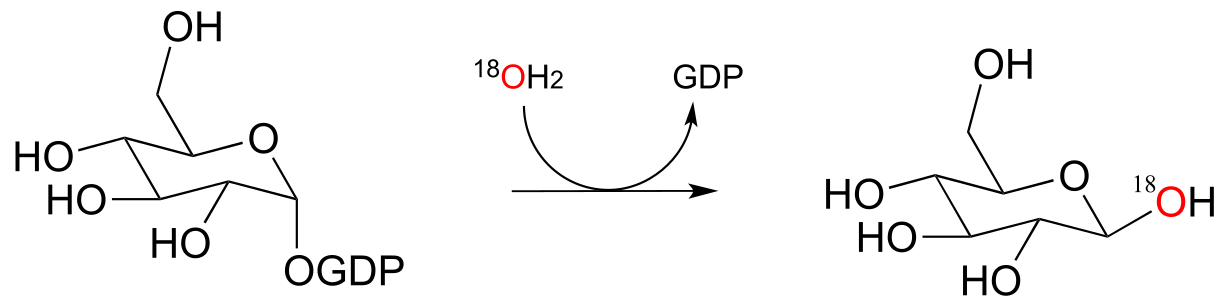
a) The researchers concluded that the reaction was not a phosphate group transfer. Explain their reasoning.
b) Draw the products that would be expected if the reaction actually did proceed by a phosphate transfer mechanism (be sure to show stereochemistry and the location of the 18O atom). (Biochemistry 2000, 39, 8603)
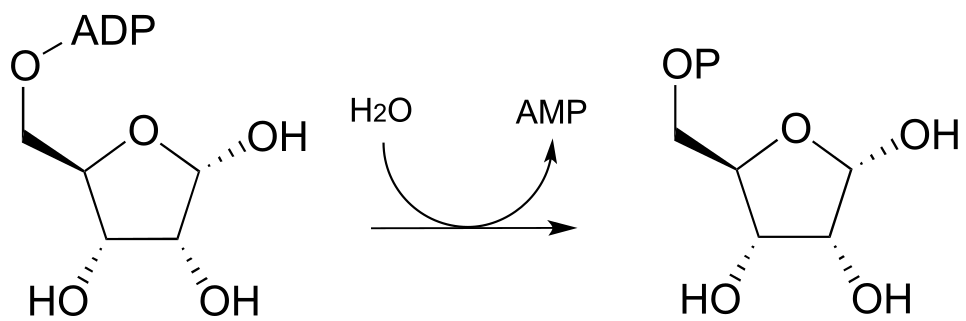
P9.8: The reaction below proceeds with a direct attack by a water molecule on the substrate, but the hydrolysis could be expected to proceed through two possible mechanisms. Draw two possible mechanisms for the reaction run in H218O. Trace the progress of the 18O ‘label’ throughout each mechanism to see where it ends up: this should indicate to you how the two mechanisms could be (and in fact were!) distinguished experimentally.
P9.9: Glucose-6-phosphate is dephosphorylated to glucose in the last step of the gluconeogenesis pathway (EC 5.3.1.9). The reaction is not a direct hydrolysis: like the phosphotyrosine phosphatase reaction we saw in this chapter it involves formation of a phosphoenzyme intermediate, but in this case the enzyme residue acting as the initial phosphate acceptor is an active site histidine rather than an asparate. Given this information, propose a likely mechanism for the reaction.
P9.10: (This question assumes a basic knowledge of DNA structure and the idea of supercoiling). DNA topoisomerase enzymes catalyze the temporary ‘nicking’ of one strand of double-stranded DNA, which allows supercoiled DNA to ‘unwind’ before the nicked strand is re-ligated. During the unwinding process, the 5’ end of the nicked strand is transferred to a tyrosine in the enzyme’s active site, effectively holding it in place while the 3’ end rotates. Overall, the stereochemical configuration of the bridging phosphate is retained. Propose a likely mechanism for this nicking and re-ligating process. (Biochemistry 2005, 44, 11476.)
P9.11: Pictured below is a series of phosphate group transfer steps in the early part of isoprenoid biosynthesis in bacteria. With the knowledge that the atoms in green are derived from ATP, predict the structures of compounds A, B and C. (EC 2.7.7.60, EC 2.7.1.148, EC 4.6.1.12)
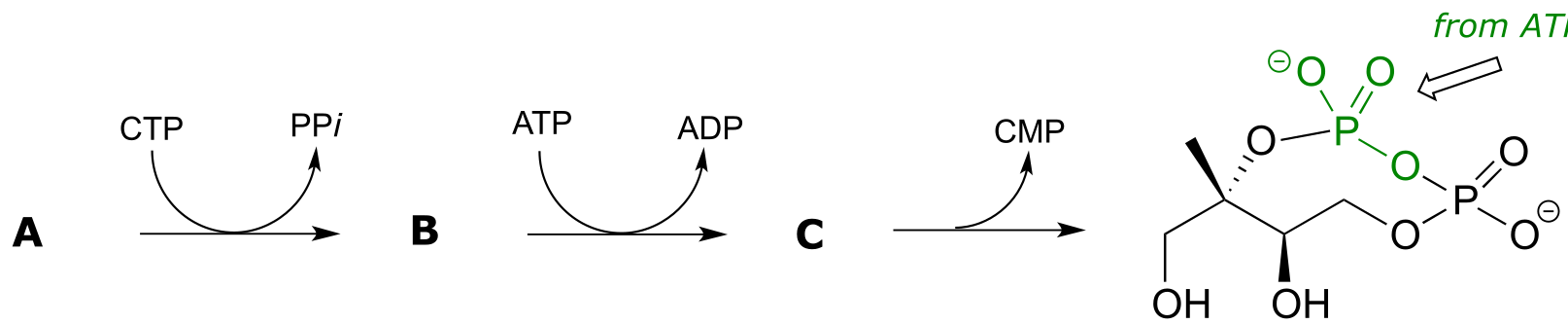
P9.12: The reaction below shows the synthesis of glucose-UDP, an important intermediate in carbohydrate biosynthesis. Notice that UTP (instead of ATP) is the phosphate donor. Identify the by-product denoted below by a question mark.
Molecules and Cells 2010, 29, 397; E.C. 2.7.7.9
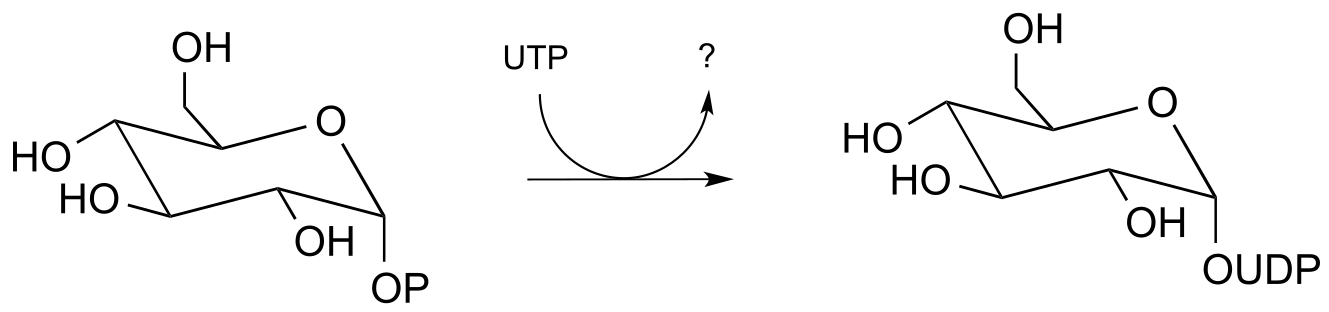
P9.13: Isomerization of 3-phosphoglycerate to 2-phosphoglycerate (EC 5.4.2.1, a reaction in glycolysis) has been shown to occur with the participation of a phosphohistidine residue in the enzyme’s active site. The two phosphate groups are distinguished in the figure below by color. With this information, propose a mechanism for the reaction. (J. Mol. Biol. 1999, 286, 1507
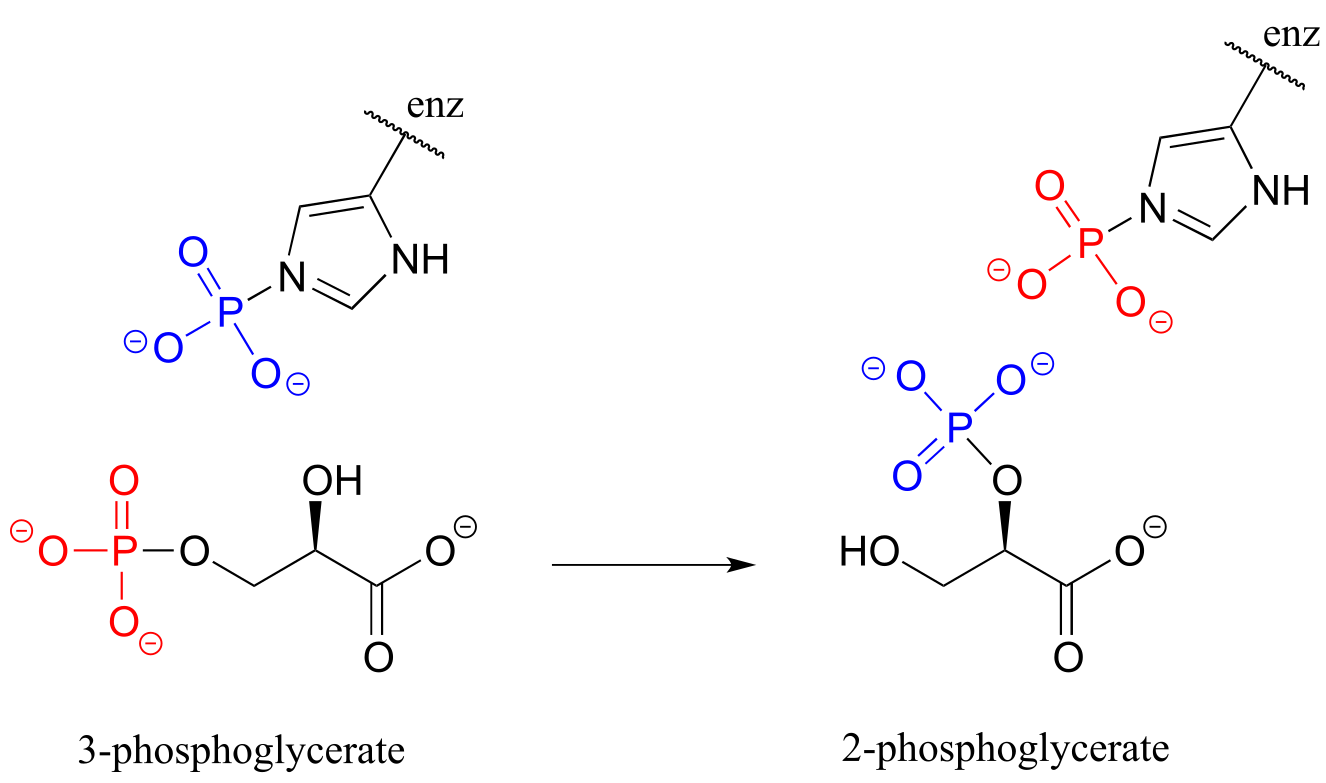
P9.14: The gluconeogenesis (sugar-building) pathway enzyme glucose-6-phosphatase catalyzes an indirect phosphate hydrolysis reaction with a phosphohistidine intermediate (‘indirect hydrolysis’ in this context means that a water molecule does not directly attack glucose-6-phosphate).
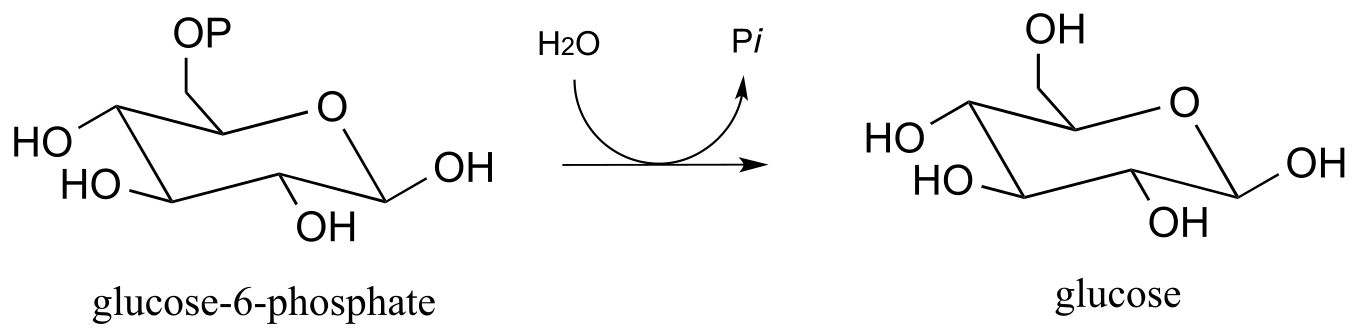
Researchers wanted to confirm that the hydrolysis in this reaction is indirect, rather than direct. It turns out that the same enzyme is also capable of catalyzing the transfer of the phosphate group from glucose-6-phosphate to the hydroxyl group on carbon #6 of another glucose molecule (instead of to water, which is the natural reaction). The enzyme-catalyzed transfer of phosphate between two glucose substrates is reversible.
The researchers incubated the enzyme with labeled glucose-6-phosphate, in which in the phosphate center was chiral (with the R configuration) due to the incorporation of 17O and 18O isotopes. They also included a high concentration of glucose in the reaction mixture, which ensured that the glucose-to-glucose transfer reaction predominated and hydrolysis (the ‘natural’ reaction) did not take place. After allowing the reaction to reach equilibrium, they isolated the glucose-6-phosphate and looked at the configuration of the phosphate group.
Given what you have just learned about the enzyme mechanism, predict what the researchers found in this experiment, explain your prediction, and draw the appropriate structure(s), including stereochemistry. Assume that the glucose-to-glucose mechanism is identical to the hydrolysis mechanism, aside from the identity of the ultimate phosphate acceptor. Biochem J., 1982*, 201,* 665*.*