15: Oxidation and reduction reactions
Contents
15: Oxidation and reduction reactions#

photo credit: Johann Schwarz (https://www.flickr.com/photos/blacksportsphotos/)
Introduction
Theo Ross was not doing very well at his summer job, and he was frustrated. His boss had given him specific instructions, and yet Theo kept botching the job, over and over again. Theo was not used to failure – he had achieved almost perfect scores on both the ACT and SAT college entrance exams, and was headed to Stanford University in the fall. Why couldn’t he get it right? It wasn’t brain surgery, after all.
Well, actually – it was brain surgery.
An April 17, 2014 article in Sports Illustrated tells Theo’s story. Wanting to do something interesting over the summer of 2010 before he started college, Theo had applied for a research internship at the National Institutes of Health in Bethesda, Maryland. This was an extremely competitive program normally reserved for outstanding college students, but somehow Theo had managed to win a coveted spot in the program, working with Dr. Dorian McGavern, a neurologist studying how meningitis effects the brain. Dr. McGavern assigned Theo the task of performing ‘skull-thinning’ surgery on mice, part of which involved using a special saw to shave down a small section of the bone in order to gain access to the brain. It was a delicate procedure, something that even some experienced neurosurgeons who had tried it had found challenging. Any small slip resulted in a concussion to the mouse’s brain, rendering it useless for the study. Theo just couldn’t get the hang of it, and ended up concussing one mouse after another.
You have probably heard the old expression: “when life gives you lemons, make lemonade”. Theo made a lot of lemonade that summer.
Theo and Dr. McGavern eventually realized that his failure at the procedure actually presented an opportunity to observe what happens to a brain right after a concussive injury. Theo started doing more skull-thinning surgeries, but now the goal was to cause concussion, rather than to avoid it. The concussed mice (who had been anaesthetized prior to the surgery) were immediately strapped under a microscope so that Theo could observe how their brains responded to the injury. This was new, and very exciting stuff: most of what neurologists knew about concussions up to that point had come from MRI (magnetic resonance imaging – see chapter 5) or autopsies. Nobody knew very much about what happens at the cellular level in a brain in the minutes and hours after a concussion has occurred. In addition, the problem of traumatic head injuries and the long-lasting effects they cause was becoming an increasingly hot topic in the news, critically relevant to thousands of veterans returning from Iraq and Afghanistan as well as to football players and other athletes in contact sports– including Theo, who had been a competitive wrestler in high school. (You might have been wondering why Theo’s story appeared in Sports Illustrated – now you know.)
Theo spent the rest of that summer, and every spring break and summer vacation over the next few years, working in McGavern’s lab on the new project. He and McGavern found evidence that the ‘hidden’ damage to a concussed brain – that which went undetected in MRI scans but could come back to haunt the victim years later in the form of recurring headaches, memory loss, and depression – may be caused by a type of molecule referred to as ‘reactive oxygen species’, or ROS, leaking from damaged tissues into the brain. ROS are potentially harmful byproducts of respiration such as hydrogen peroxide (H2O2) that are constantly being produced in our cells. Although ROS can cause serious oxidative damage if they are allowed to build up, our bodies have evolved ways to deal with them, using so-called ‘ROS scavengers’ to convert them to something innocuous like water.
With this new understanding, Theo and his mentor had another idea: what if they could prevent the ROS from causing further damage to a recently concussed brain by applying an scavenger to the injury? After some trial and error, they found that an ROS scavenger compound called glutathione, when applied directly to the skull of a concussed mouse within a few minutes to three hours after the injury, could permeate the bone and react with the ROS. Brain cells from these glutathione-treated mice appeared normal, with none of the signs of ROS damage Theo was used to seeing.
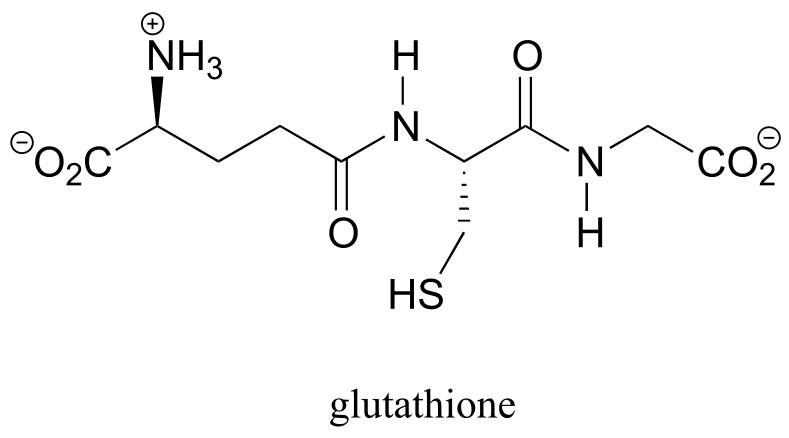
fig 1a
The road from an initial scientific discovery to a safe and effective medical treatment is often a very long one, but Theo Roth and Dorian McGavern appear to have made a discovery that could eventually help prevent some of the most devastating and long-term damage caused by traumatic head injuries. In the end, it’s a very good thing that Theo’s hands were not cut out for brain surgery.
***
The chemistry of oxidation and reduction - often called ‘redox’ chemistry - is central to Theo Roth’s discovery about what happens to a concussed brain at the molecular level. This chapter is dedicated to redox chemistry. We’ll begin with a reminder of what you learned in General Chemistry about the fundamentals of redox reactions in the context of inorganic elements such as iron, copper and zinc: reduction is a gain of electrons, and oxidation is a loss of electrons. Then, we’ll expand our understanding to include bioorganic redox reactivity, examining among other things how alcohols are converted to ketones and aldehydes, aldehydes are converted to carboxylic acids, and amines are converted to imines. We will also talk about redox reactions in the broader context of metabolism in living things.
A central player in some of the biochemical redox reactions we will see is the coenzyme called glutathione, Theo Roth’s ‘magic bullet’ molecule that was able to rescue mouse brain cells from death by oxidation. We’ll see how glutathione acts as a mediator in the formation and cleavage of disulfide bonds in proteins, and how it acts as an ‘ROS scavenger’ to turn hydrogen peroxide into water.
**
**
Section 15.1: Oxidation and reduction of organic compounds - an overview#
You are undoubtedly already familiar with the general idea of oxidation and reduction: you learned in general chemistry that when a compound or element is oxidized it loses electrons, and when it is reduced it gains electrons. You also know that oxidation and reduction reactions occur in tandem: if one species is oxidized, another must be reduced at the same time - thus the term ‘redox reaction’.
Most of the redox reactions you have seen previously in general chemistry probably involved the flow of electrons from one metal to another, such as the reaction between copper ion in solution and metallic zinc:
Cu+2(aq) + Zn(s) → Cu(s) + Zn+2(aq)
Exercise 15.1: Reading the reaction above from left to right, which chemical species is being oxidized? Which is being reduced?
When we talk about the oxidation and reduction of organic compounds, what we are mainly concerned with is the number of carbon-heteroatom bonds in the compound compared to the number of carbon-hydrogen bonds. (Remember that the term ‘heteroatom’ in organic chemistry generally refers to oxygen, nitrogen, sulfur, or a halogen).
Oxidation of an organic compound results an increase in the number of carbon-heteroatom bonds, and/or a decrease in the number of carbon-hydrogen bonds.
Reduction of an organic compound results in a decrease in the number of carbon-heteroatom bonds, and/or an increase in the number of carbon-hydrogen bonds.
Below are a number of common functional group transformations that are classified as redox.
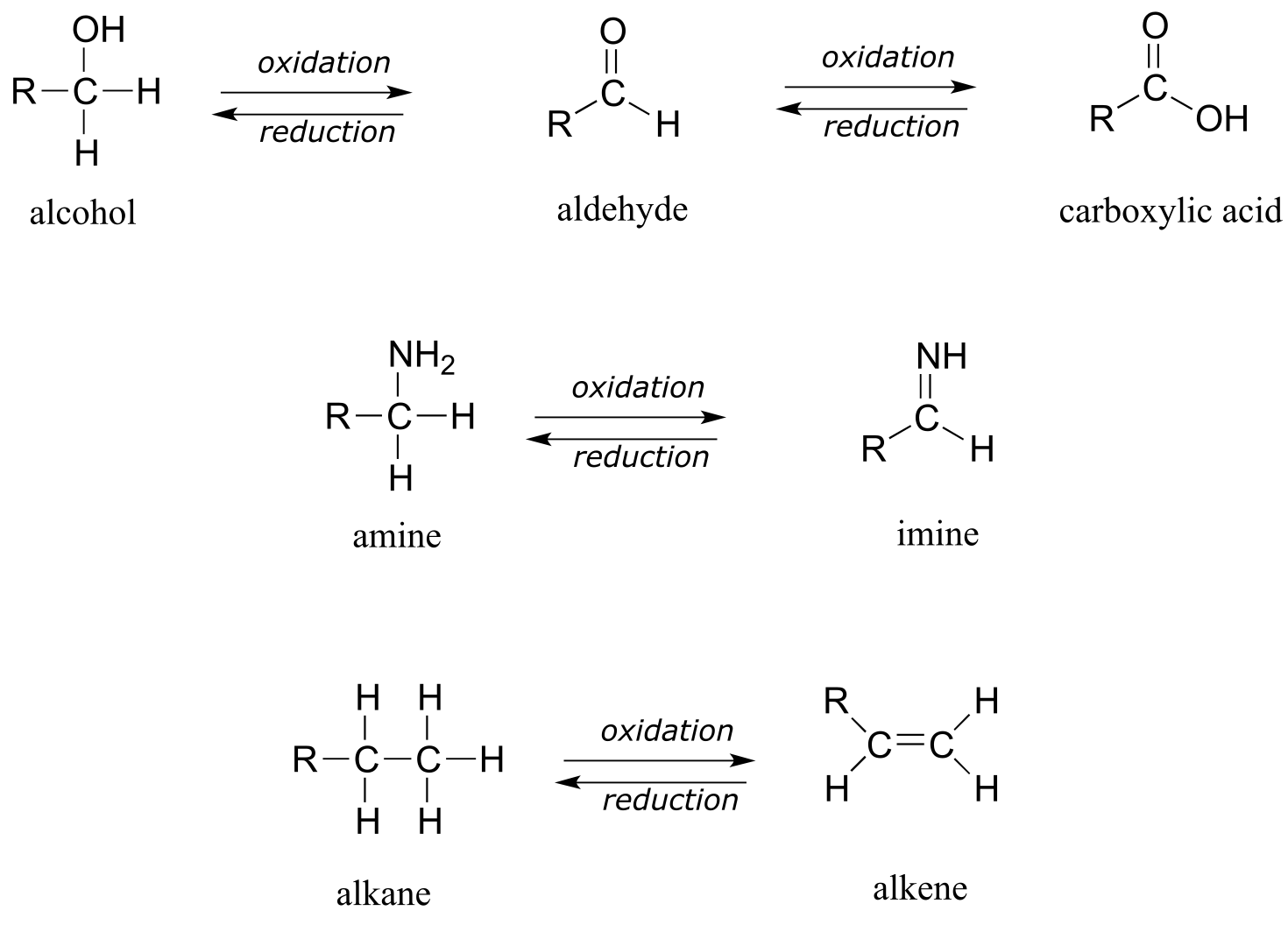
fig1
Heteroatoms such as oxygen and nitrogen are more electronegative than carbon, so when a carbon atom gains a bond to a heteroatom, it loses electron density and is thus being oxidized. Conversely, hydrogen is less electronegative than carbon, so when a carbon gains a bond to a hydrogen, it is gaining electron density, and thus being reduced.
Exercise 15.2: The hydration of an alkene to an alcohol is not classified as a redox reaction. Explain.
For the most part, when talking about redox reactions in organic chemistry we are dealing with a small set of very recognizable functional group transformations. The concept of oxidation state can be useful in this context. When a compound has lots of carbon-hydrogen bonds, it is said to be in a lower oxidation state, or a more reduced state. Conversely, if it contains a lot of carbon-heteroatom bonds, it is said to be in a higher oxidation state.
We’ll start with a series of single carbon compounds as an example. Methane, in which the carbon has four bonds to hydrogen, is the most reduced member of the group. The compounds become increasingly oxidized as we move from left to right, with each step gaining a bond to oxygen and losing a bond to hydrogen. Carbon dioxide, in which all four bonds on the carbon are to oxygen, is in the highest oxidation state.
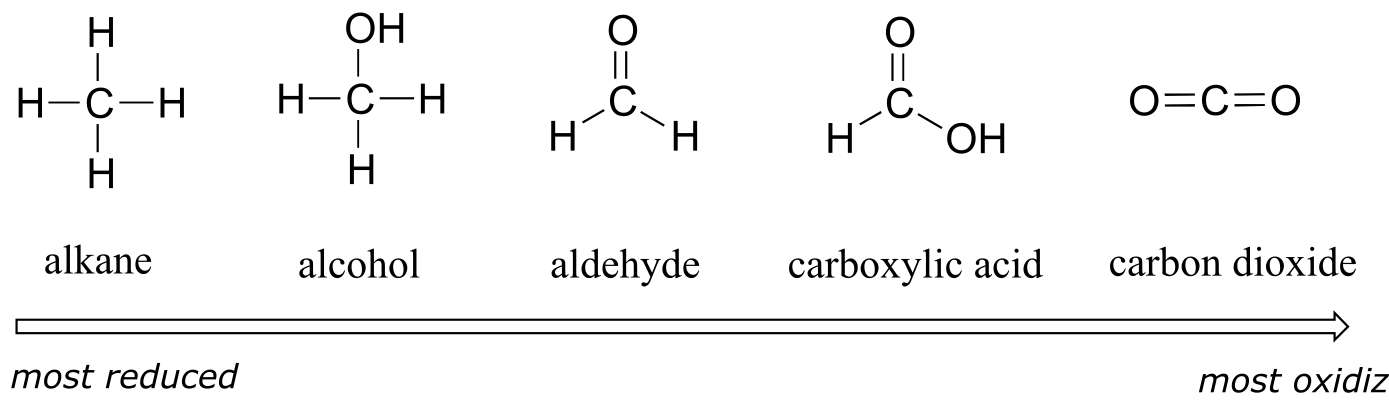
fig 3
More generally, we can rank the oxidation state of common functional groups:

fig 4
The alkane oxidation state is the most reduced. Alcohols, thiols, amines, and alkenes are all at the same oxidation state: therefore, a reaction converting one of these groups to another - an alcohol to alkene conversion, for example - is not a redox reaction. Aldehydes, however, are at a higher oxidation state than alcohols, so an alcohol to aldehyde conversion is an oxidation. Likewise, an imine to amine conversion is a reduction, but an imine to ketone conversion is not a redox reaction.
It is important to keep in mind that oxidation and reduction always occurs in tandem: when one compound is oxidized, another compound must be reduced. Often, organic chemists will use the terms oxidizing agent and reducing agent to refer to species that are commonly used, by human chemists or by nature, to achieve the oxidation or reduction of a variety of compounds. For example, chromium trioxide (CrO3) is a laboratory oxidizing agent used by organic chemists to oxidize a secondary alcohol to a ketone, in the process being reduced to H2CrO3. Sodium borohydride (NaBH4) is a laboratory reducing agent used to reduce ketones (or aldehydes) to alcohols, in the process being oxidized to NaBH3OH.
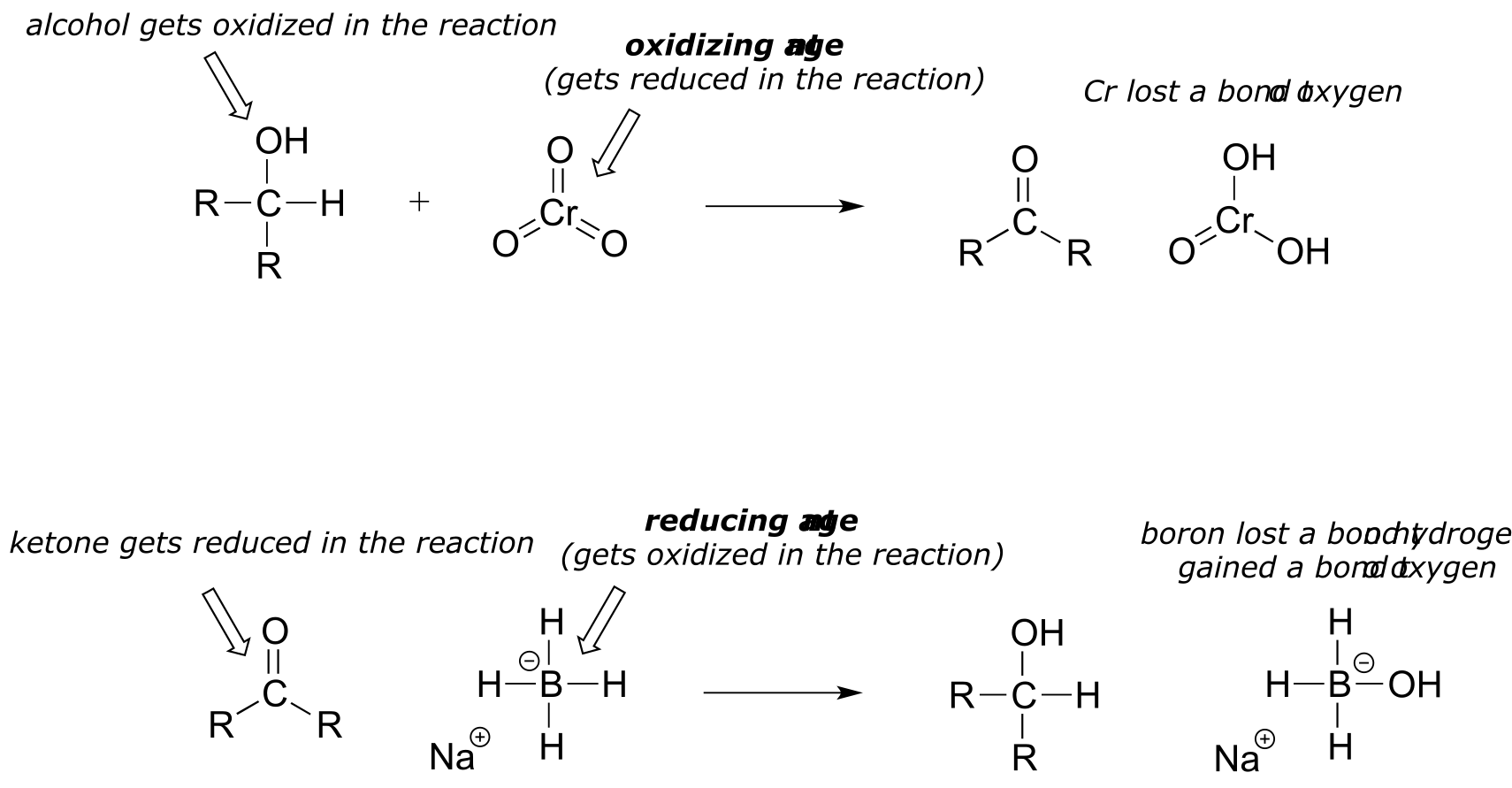
There is a wide selection of oxidizing and reducing agents available for use in the organic chemistry laboratory, each with its own particular properties and uses. For example, while sodium borohydride is very useful for reducing aldehyde and ketone groups to alcohols, it will not reduce esters and other carboxylic acid derivatives. If you take a course in synthetic organic chemistry, you will learn about the use of many of these agents.
In this book, of course, we are concerned primarily with the organic chemistry that occurs within a living cell. A large part of this chapter will be spent looking at the action of two very important classes of coenzymes - the nicotinamides and the flavins - that serve as biochemical oxidizing and reducing agents. We also consider the oxidation and reduction of sulfur atoms in thiol groups, especially the thiol group on the side chain of cysteine residues in proteins.
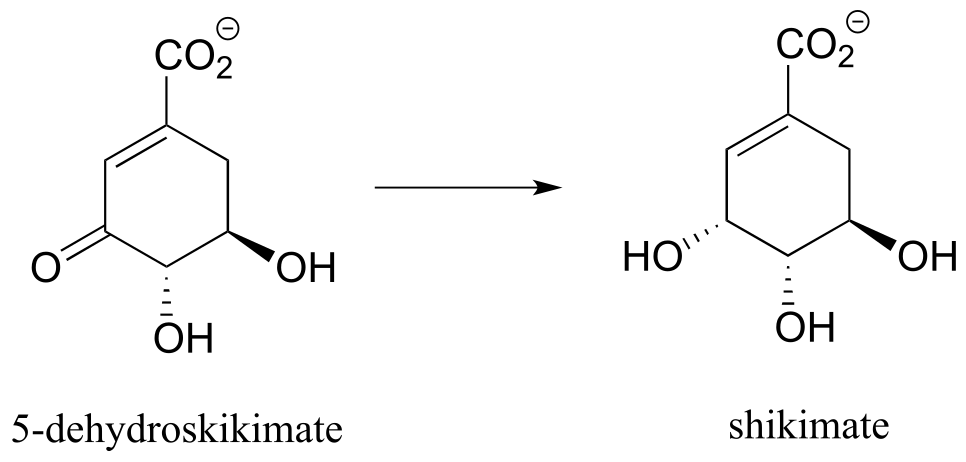
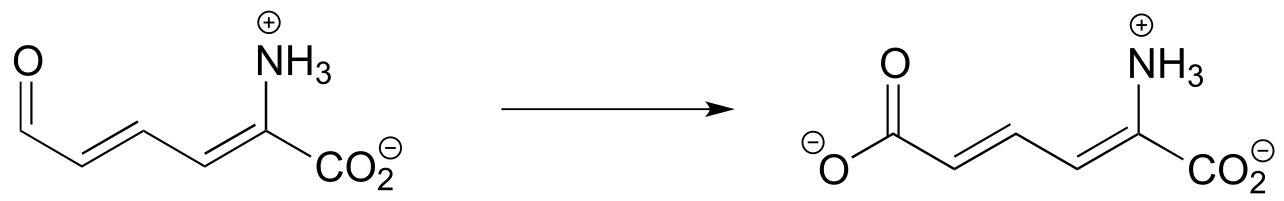
Exercise 15.3: Each of the biochemical transformations shown below is a step in amino acid metabolism. For each, state whether the substrate is being oxidized, reduced, or neither oxidized nor reduced.
a) (from aromatic amino acid biosynthesis)
b) (from the biosynthesis of arginine and proline)
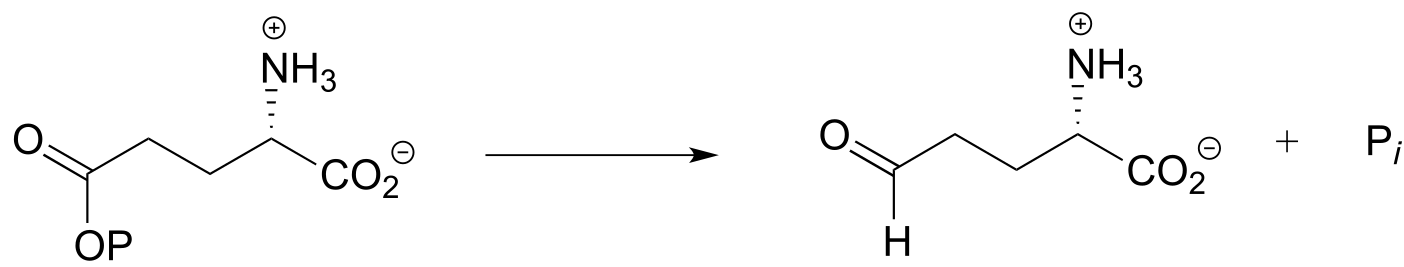
c) (from the catabolism of lysine)

d) (from the catabolism of tryptophan)
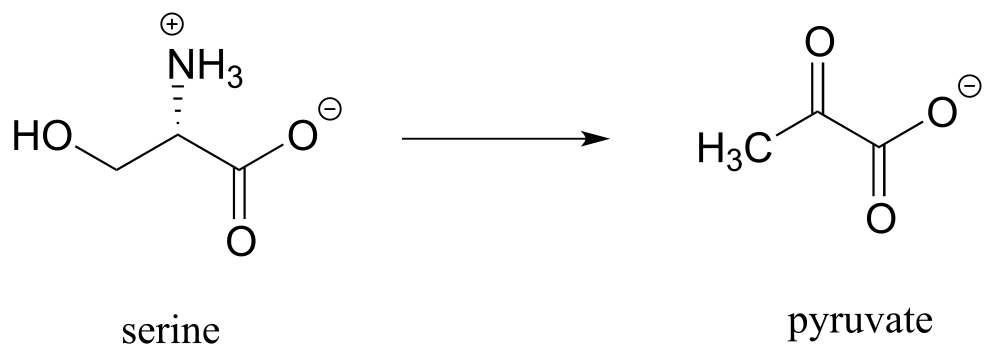
e) (from the catabolism of serine)
Section 15.2: Oxidation and reduction in the context of metabolism#
Think back again to the redox chemistry that you learned in your general chemistry course. A common experiment in a general chemistry lab is to set up a galvanic cell consisting of a copper electrode immersed in an aqueous copper nitrate solution, connected by a wire to a zinc electrode immersed in an aqueous zinc nitrate solution.
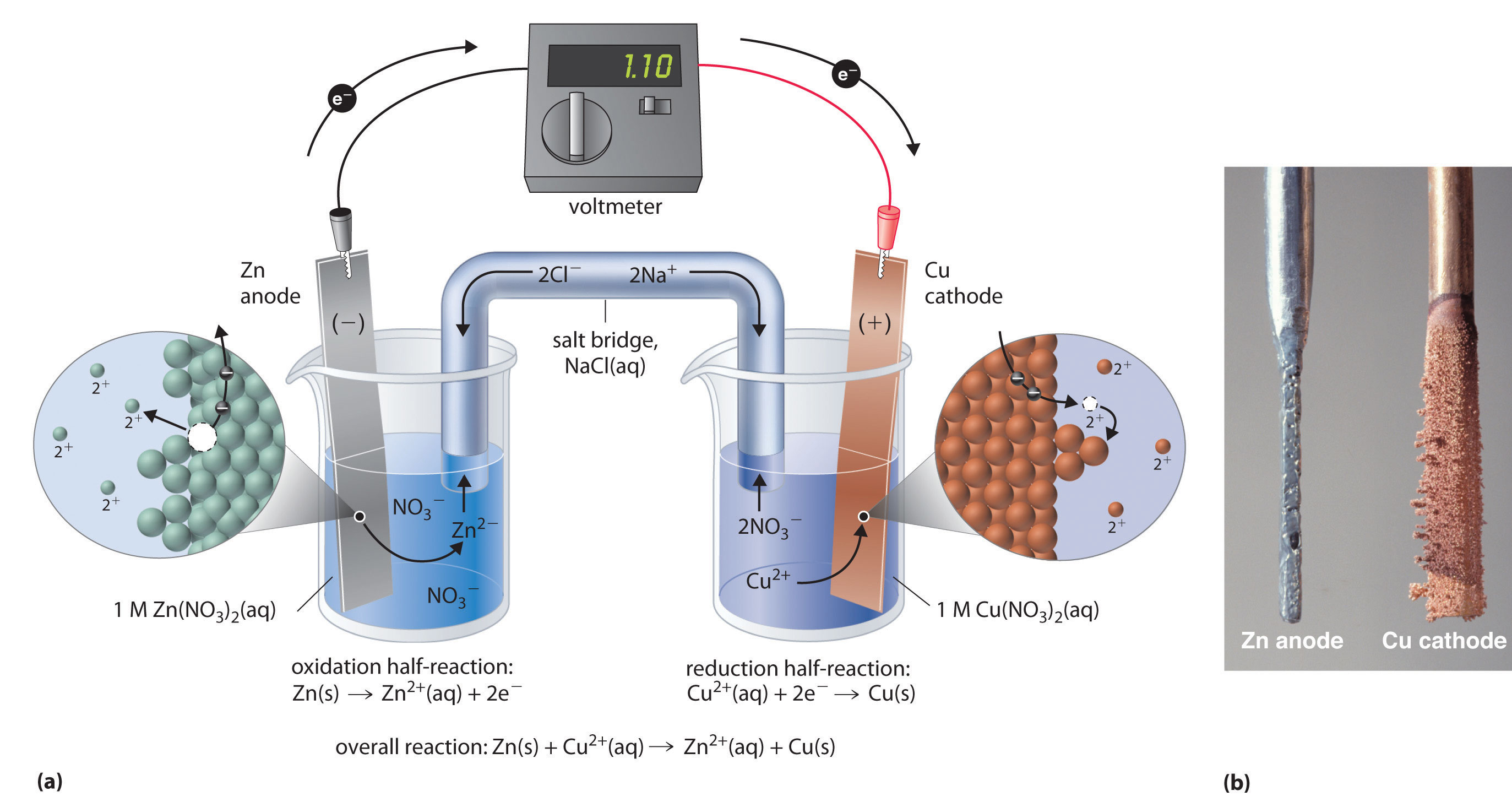
(http://chemwiki.ucdavis.edu)
When the cell is completed with a salt bridge, an electrical current begins to flow - what we have is a simple battery (figure a above). Over time, the copper electrode gets heavier as metallic copper is deposited on the copper cathode, while the zinc anode slowly dissolves into solution (figure b above). The redox reaction occurring here is:
Cu+2(aq) + Zn(s) → Cu(s) + Zn+2(aq) + energy
Electrons flow from zinc metal to copper cations, creating zinc cations and copper metal: in other words, zinc metal is being oxidized to zinc cation and copper cation is being reduced to copper metal, as expressed by the two relevant half-cell reactions:
Cu+2(aq) + 2e- → Cu0(s)
Zn0(s) → Zn+2(aq) + 2e-
We can predict before we set up the cell that the spontaneous flow of electrons will go in the zinc to copper direction, just by looking at a table of standard reduction potentials (such a table was no doubt in your general chemistry text).
Standard reduction potentials at 25 oC
| Reduction half-reaction | Reduction potential (volts) |
|---|---|
| Ag+1(aq) + e- → Ag0(s) | 0.800 |
| Cu+2(aq) + 2e- → Cu0(s) | 0.337 |
| H+1(aq) + 2e- → H2(g) | 0 (standard) |
| Pb+2(aq) + 2e- → Pb0(s) | -0.126 |
| Fe+2(aq) + 2e- → Fe0(s) | -0.441 |
| Zn+2(aq) + 2e- → Zn0(s) | -0.763 |
Copper ion (Cu+2) has a higher standard reduction potential than zinc ion (Zn+2), meaning that, under identical conditions, more energy is released by reducing one mole of Cu+2 ion to Cu0 metal than is released by reducing one mole of Zn+2 ion to Zn0 metal.. Another way to think about this is to imagine that the copper ion ‘wants’ to gain electrons more than the zinc ion does*.* Conversely, zinc metal ‘wants’ to lose electrons more than the copper metal does. Therefore, transfer of two electrons from zinc metal to Cu2+ is a thermodynamically downhill process, whereas the reverse process - transfer of two electrons from copper metal to Zn2+ - is thermodynamically uphill.
Cu+2(aq) + Zn(s) → Cu(s) + Zn+2(aq) + energy
Cu(s) + Zn+2(aq) + energy → Cu+2(aq) + Zn(s)
Let’s now extend the idea of redox reactions to the context of metabolism in living things. When we ‘burn’ glucose for energy, we transfer (by a series of enzyme-catalyzed reactions) electrons from glucose to molecular oxygen (O2), oxidizing the six carbon molecules in glucose to carbon dioxide and at the same time reducing the oxygen atoms in O2 to water. The overall chemical equation is:
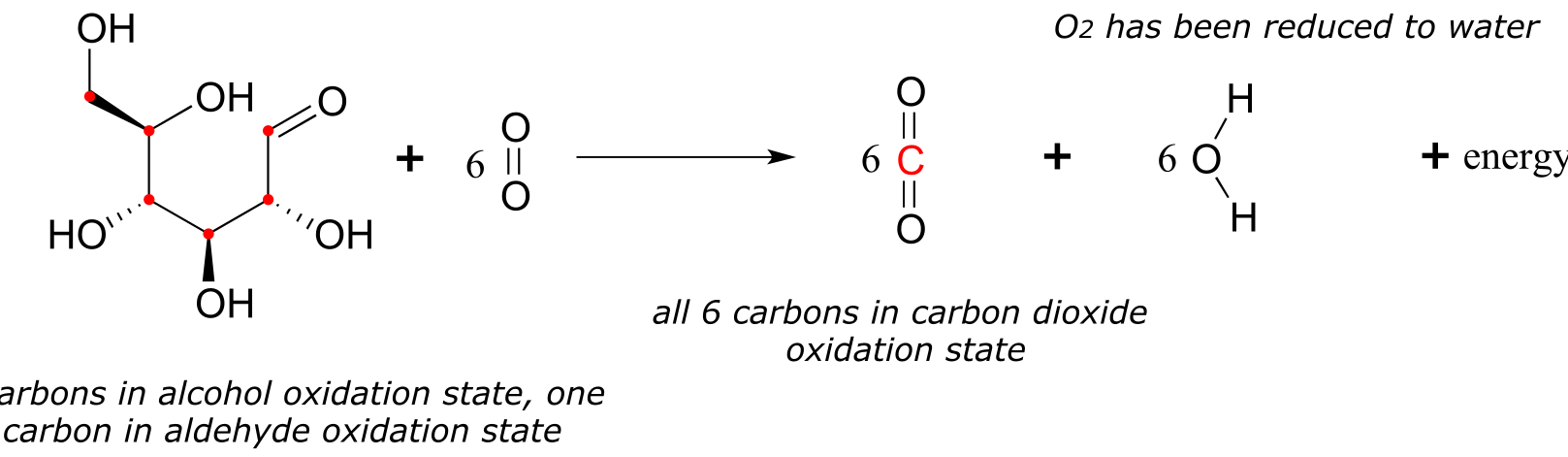
fig 8
The transfer of electrons from glucose to O2 is a thermodynamically downhill, energy-releasing process, just like the transfer of electrons from zinc metal to copper ion. And while you could have used the energy released by the zinc/copper redox reaction to light a small light bulb, your cells use the energy released by the glucose/oxygen redox process to carry out a wide variety of energy-requiring activities, such as walking to your organic chemistry lecture.
In your general chemistry copper/zinc experiment, was it possible to reverse the reaction so that it runs in the uphill direction - in other words, to oxidize copper and reduce zinc?
Zn+2(aq) + Cu(s) + energy → Zn(s) + Cu+2(aq)
Just ask yourself the question: is it possible to get water to flow uphill? Of course it is - but only if you supply a pump and some energy!
The same idea applies to ‘pumping’ electrons uphill in your copper-zinc electrochemical cell: all you need to do is to provide some energy in the form of an external electrical current in order to pump the electron flow in the uphill direction. You are recharging your battery.
Thinking again in a biochemical context: plants are able, by a process called photosynthesis, to reduce carbon dioxide and oxidize water to form glucose and molecular oxygen: essentially recharging the ecosystem’s biochemical battery using energy from the sun.
6CO2 + 6H2O + energy → C6H12O6 + 6O2
On a global scale, oxidation of the carbons in glucose to CO2 by non-photosynthetic organisms (like people) and the subsequent reductive synthesis of glucose from CO2 by plants is what ecologists refer to as the ‘carbon cycle’.
In general the more reduced an organic molecule is, the more energy is released when it is oxidized to CO2 . Going back to our single-carbon examples, we see that methane, the most reduced compound, releases the most energy when oxidized to carbon dioxide, while formic acid releases the least:
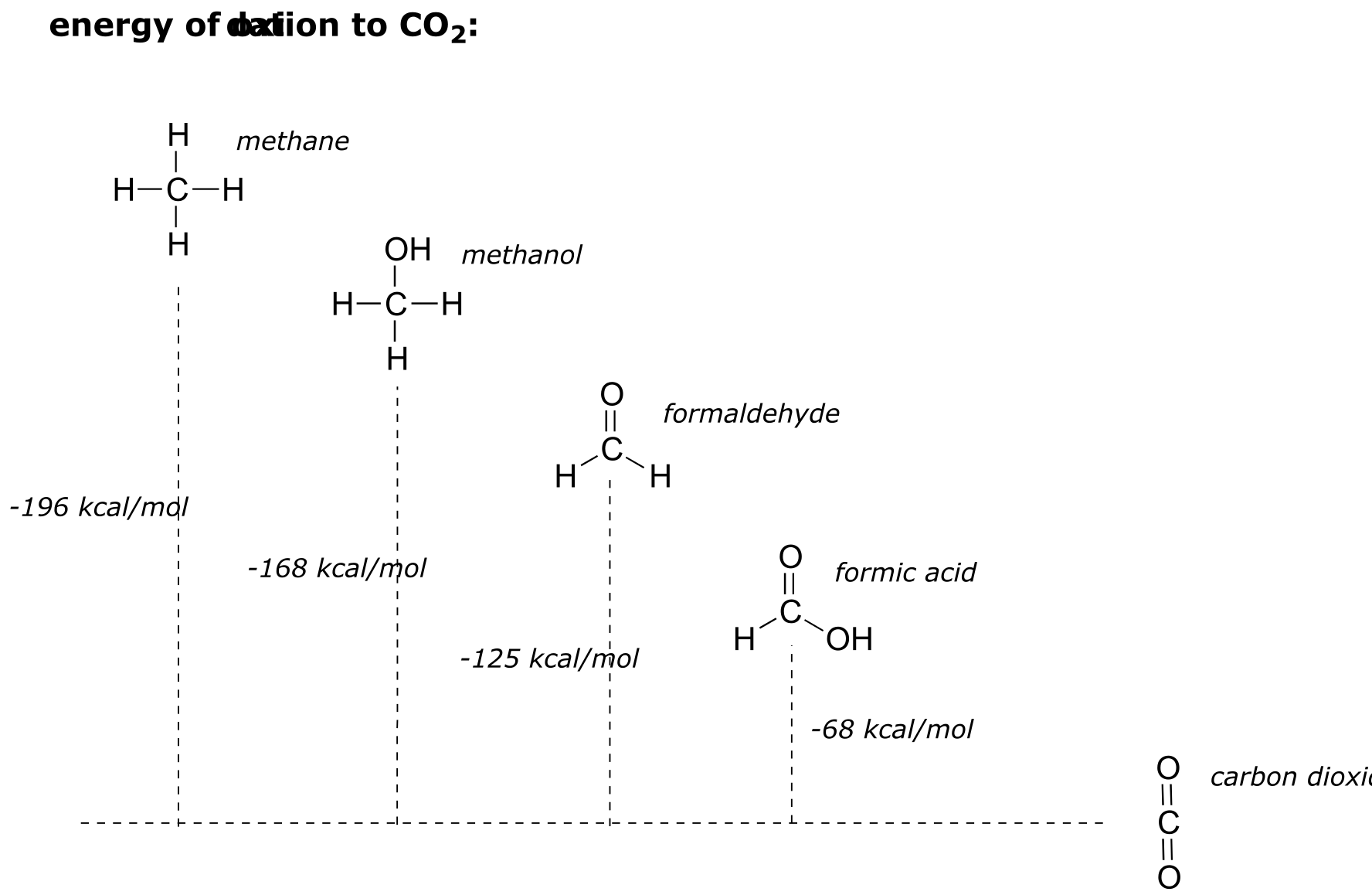
fig 9
A lipid (fat) molecule, where most of the carbons are in the highly reduced alkane state, contains more energy per gram than glucose, where five of the six carbons are in the more oxidized alcohol state (look again at the glucose structure we saw just a couple of pages back).
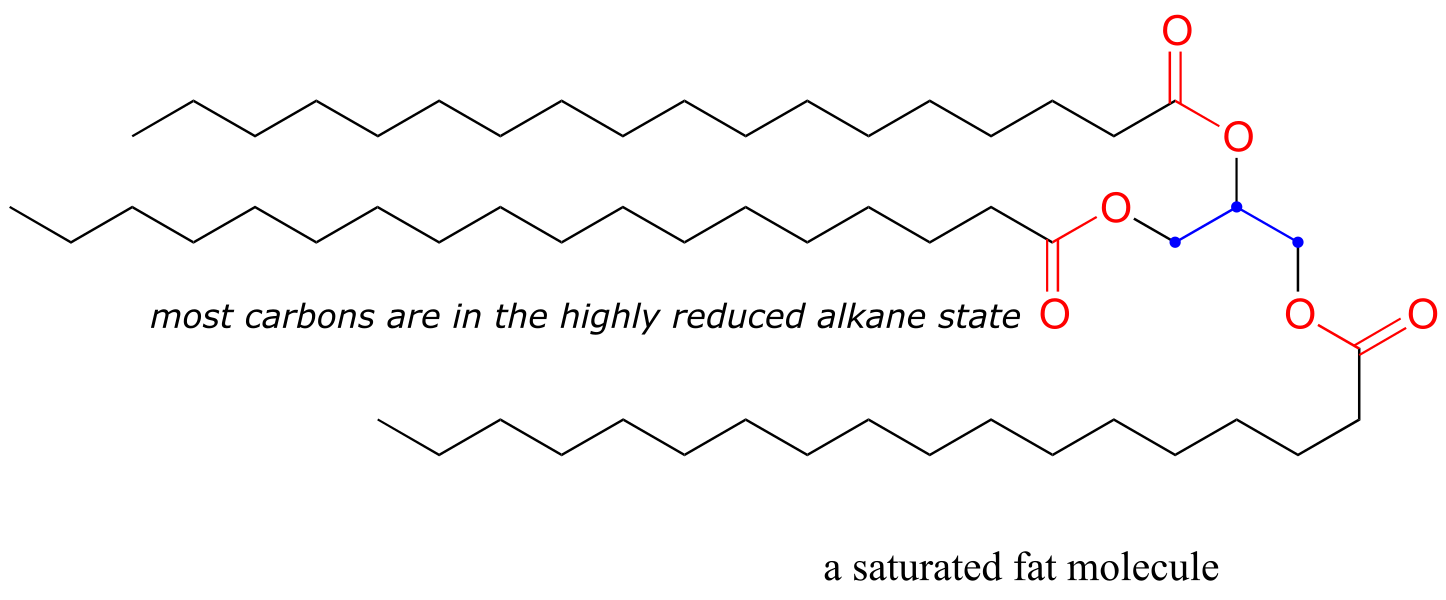
fig 10
After we break down and oxidize sugar and fat molecules to obtain energy, we use that energy to build large, complex molecules (like cholesterol, or DNA) out of small, simple precursors. Many biosynthetic pathways are reductive: the carbons in the large biomolecule products are in a reduced state compared to the small precursors. Look at the structure of cholesterol compared to that of acetate, the precursor molecule from which all of its carbon atoms are derived - you can see that cholesterol is overall a more reduced molecule.
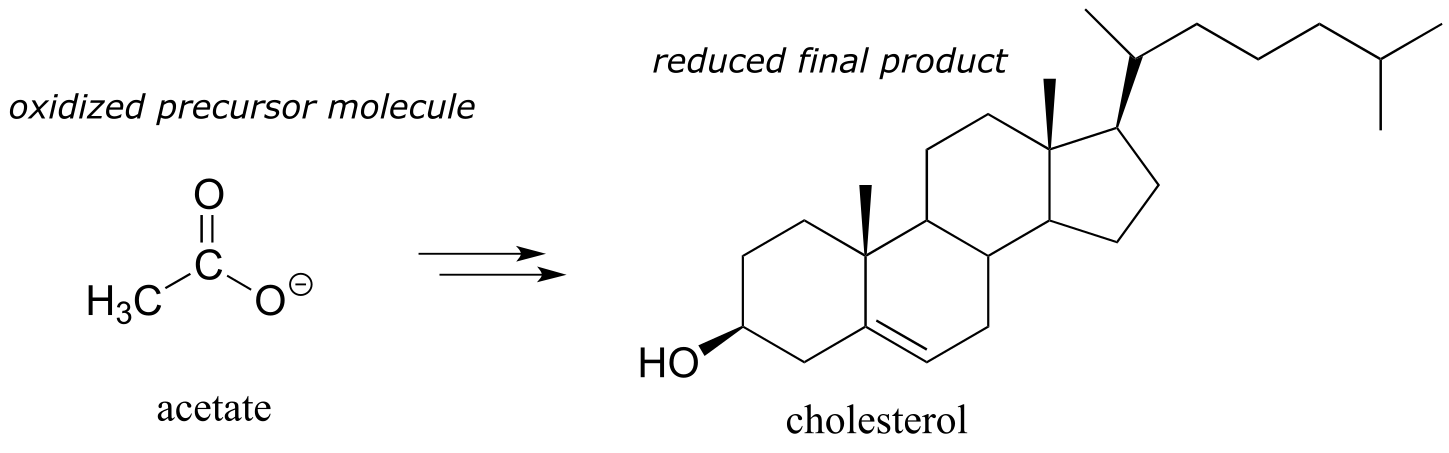
fig 11
While we are focusing here on the mechanistic details of the individual organic redox reactions involved in metabolism, if you take a course in biochemistry you will learn much more about the bigger picture of how all of these reactions fit together in living systems.
Section 15.3: Hydrogenation of carbonyl and imine groups#
Next, we’ll go on to look at the actual chemical mechanisms involved in the enzyme-catalyzed oxidation and reduction of biomolecules.
15.3A: An overview of hydrogenation and dehydrogenation#
Many of the redox reactions that you will encounter when studying the central metabolic pathways are classified as hydrogenation or dehydrogenation reactions. Hydrogenation is simply the net addition of a hydrogen (H2) molecule to a compound, in the form of a hydride ion (H-, a proton plus a pair of electrons) and a proton. Hydrogenation corresponds to reduction. Dehydrogenation reactions are the reverse process: loss of a hydride and a proton. Dehydrogenation corresponds to oxidation.
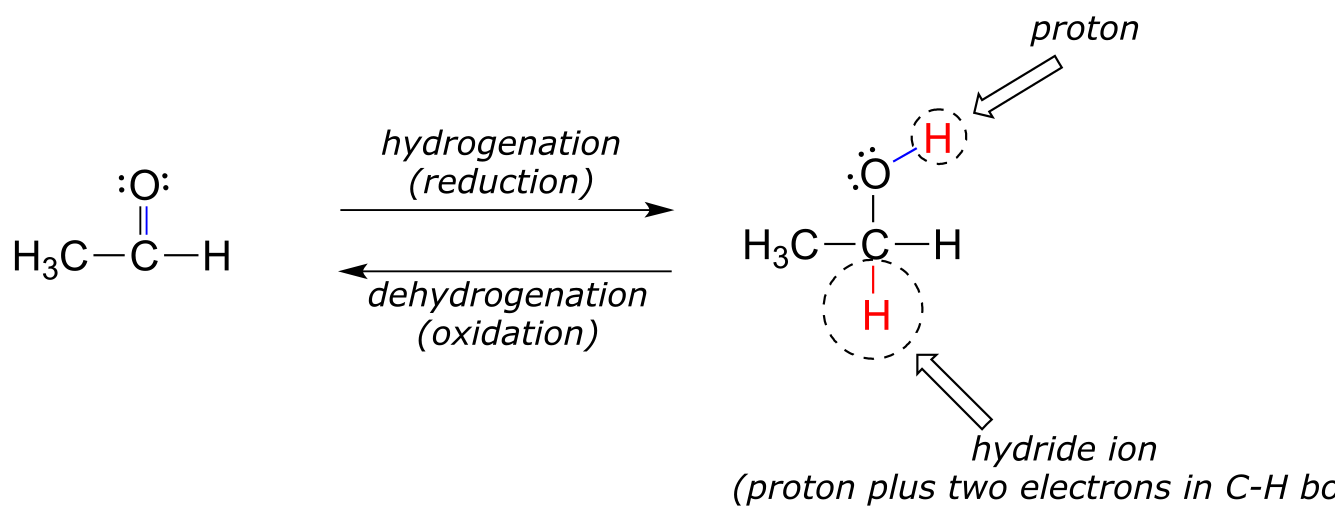
fig 2
Hydrogenation and dehydrogenation reactions can also be called hydride transfer reactions, because a hydride ion is transferred from the molecule being oxidized to the one being reduced. In the next few sections, we will learn about two important classes of coenzyme molecules that serve hydride ion acceptors (oxidizing agents) and hydride ion donors (reducing agents) in biochemical redox reactions.
Be careful not to confuse the terms hyd*rogenation and dehydrogen*ation with hydration and dehydration - the latter terms refer to the gain or loss of water, while the former terms refer to the gain or loss of hydrogen.
Many mechanistic patterns that we have already learned about in previous chapters will come into play again in this discussion, with the only variation being that here, a hydride ion will act as a nucleophile (in the hydrogenation direction) or as a leaving group (in the dehydrogenation direction). The key to understanding these reactions will be to understand how a hydride can act as a nucleophile or leaving group.
15.3B: Nicotinamide adenine dinucleotide - a hydride transfer coenzyme#
Although we are talking here about hydrides acting as nucleophiles and leaving groups, you already know that literal hydride ions are far too unstable to exist as discreet intermediates in the organic reactions of living cells (the pKa of H2, the conjugate acid of hydride, is about 35: a very weak acid, meaning hydride is a very strong base and not a reasonable species to propose for a biochemical reaction). As was alluded to earlier, biochemical hydrogenation/dehydrogenation steps require the participation of a specialized hydride transfer coenzyme. The most important of these is a molecule called nicotinamide adenine dinucleotide. The full structure of the oxidized form of this coenzyme, abbreviated NAD+, is shown below, with the active nicotinamide group colored blue. Because the redox chemistry occurs specifically at the nicotinamide ring (in blue in the figure below), typically the rest of the molecule is simply designated as an ‘R’ group.
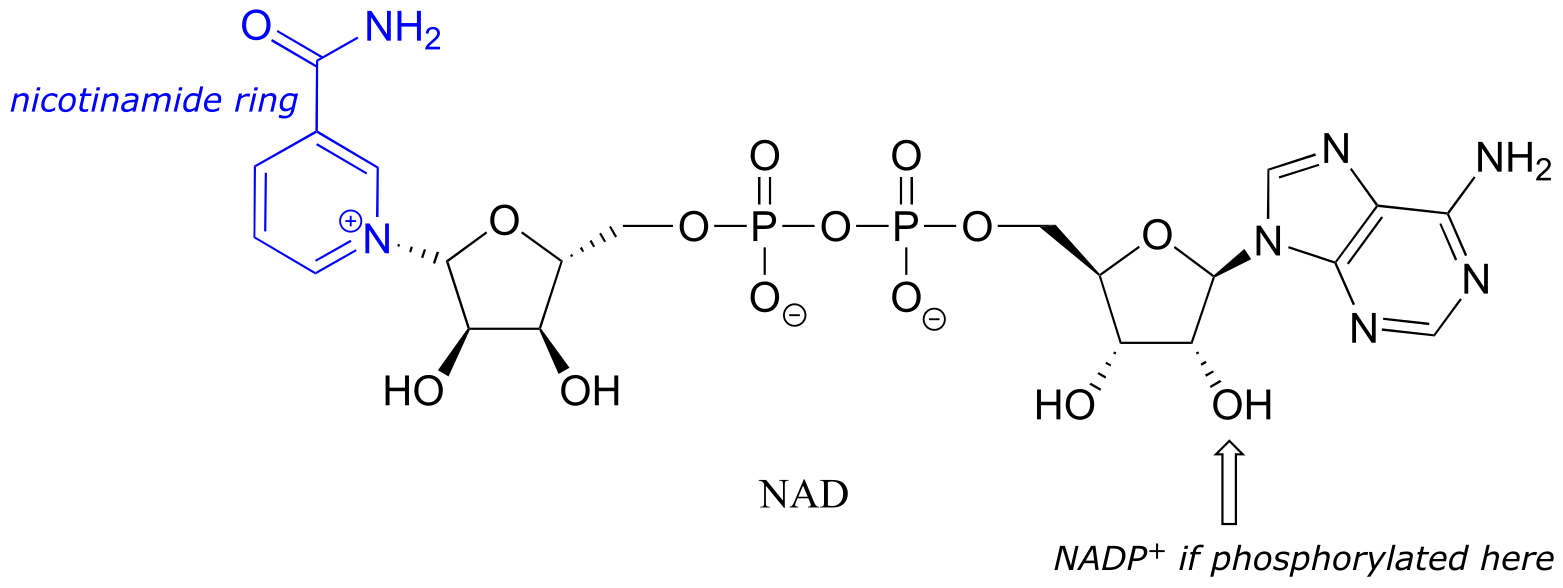
fig 16
If the hydroxyl group indicated by the arrow is phosphorylated, the coenzyme is called NADP+. The phosphate is located far from the nicotinamide ring and does not participate directly in the hydride transfer function of the cofactor. It is, however, important in a larger metabolic context: as a general rule, redox enzymes involved in catabolism (the breakdown of large molecules) typically use the non-phosphorylated coenzyme, while those involved in anabolism (biosynthesis of large molecules from small precursors) use the phosphorylated coenzyme.
NAD+ and NADP+ both function in biochemical redox reactions as hydride acceptors: that is, as oxidizing agents. The reduced forms of the coenzyme, abbreviated NADH and NADPH, serve as hydride donors: that is, as reducing agents.
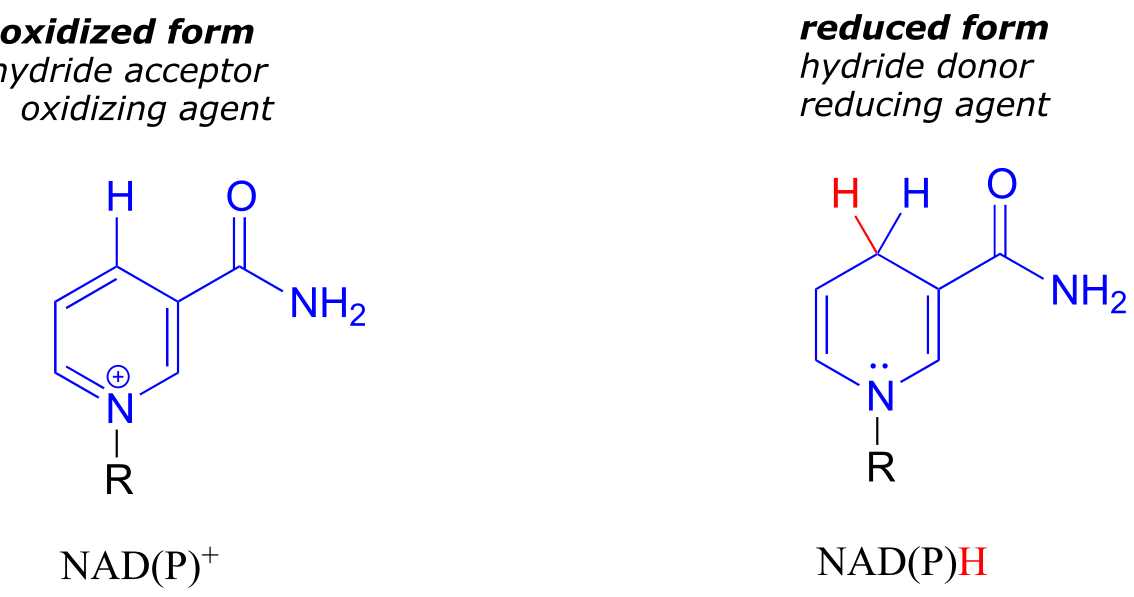
fig 5
fig 17
To understand how the nicotinamide coenzymes function in hydride transfer, let’s look at a general picture of a reversible, redox conversion from a ketone to a secondary alcohol. Mechanistically, the reaction we are about to see can be described as a nucleophilic addition to a carbonyl - a mechanism type we studied in chapter 10 - with the twist that the nucleophilic species is a hydride ion. At the beginning of the reaction cycle, both the ketone substrate and the NADH cofactor are bound in the enzyme’s active site, and carbon #4 of the nicotinamide ring is positioned very close to the carbonyl carbon of the ketone.
NAD(P)H-dependent hydrogenation (reduction) of a ketone
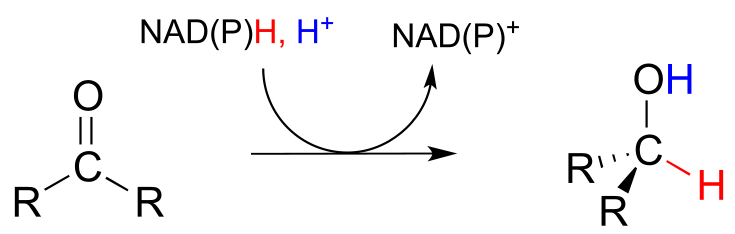
Mechanism:
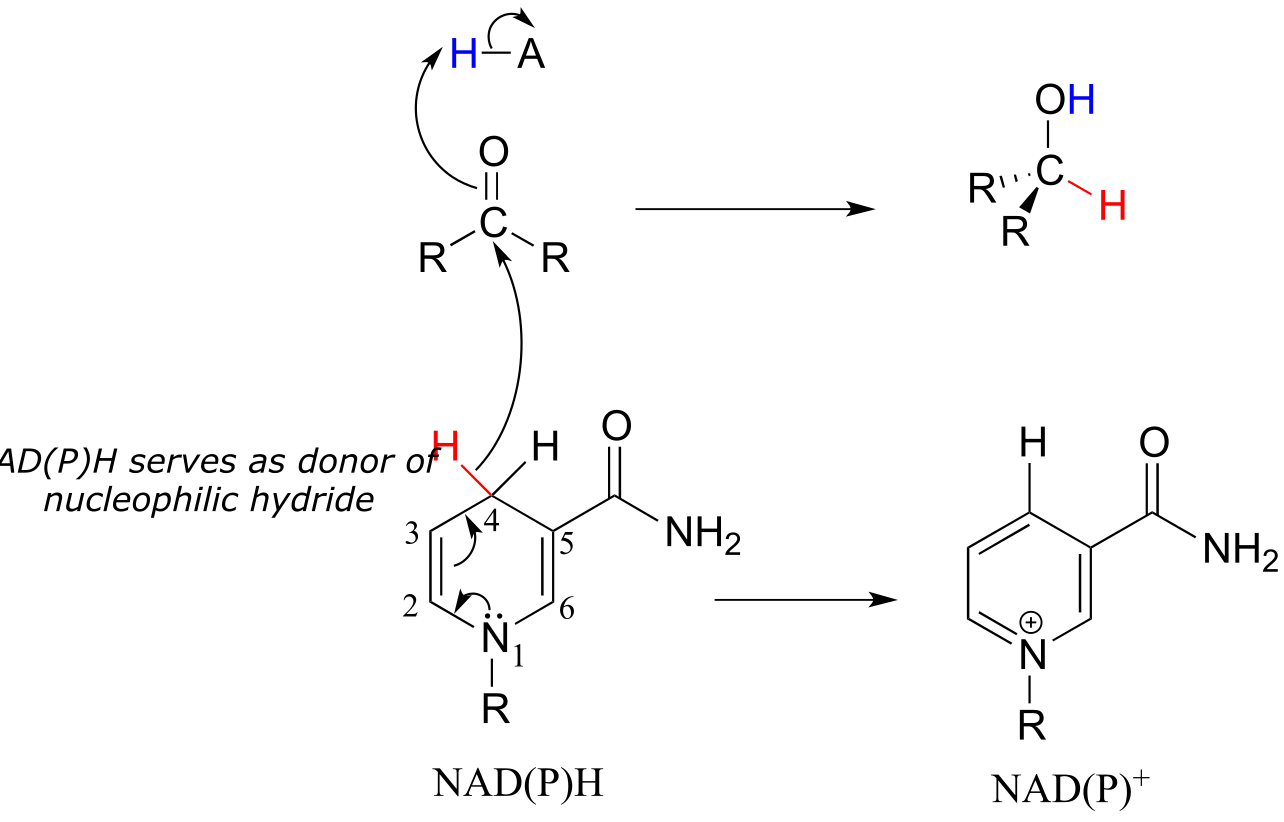
fig 18, fig 18a
As an enzymatic group transfers a proton to the ketone oxygen, the carbonyl carbon loses electron density and becomes more electrophilic, and is attacked by a hydride from NADH. Because carbon #4 of NADH is bound in such close proximity to the electrophile, this step can occur without generating a free hydride ion intermediate – the two hydride electrons can be pictured as shifting from one carbon to another. Note the products of this reaction: the ketone (which accepted a hydride and a proton) has been reduced to an alcohol, and the NADH cofactor (which donated a hydride) has been oxidized to NAD+.
The dehydrogenation of an alcohol by NAD+ is simply the reverse of a ketone hydrogenation:
NAD(P)+-dependent dehydrogenation (oxidation) of an alcohol
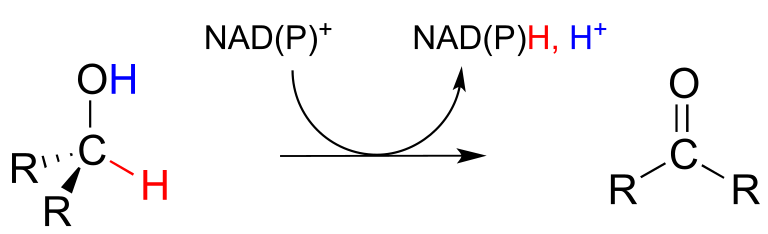
Mechanism:
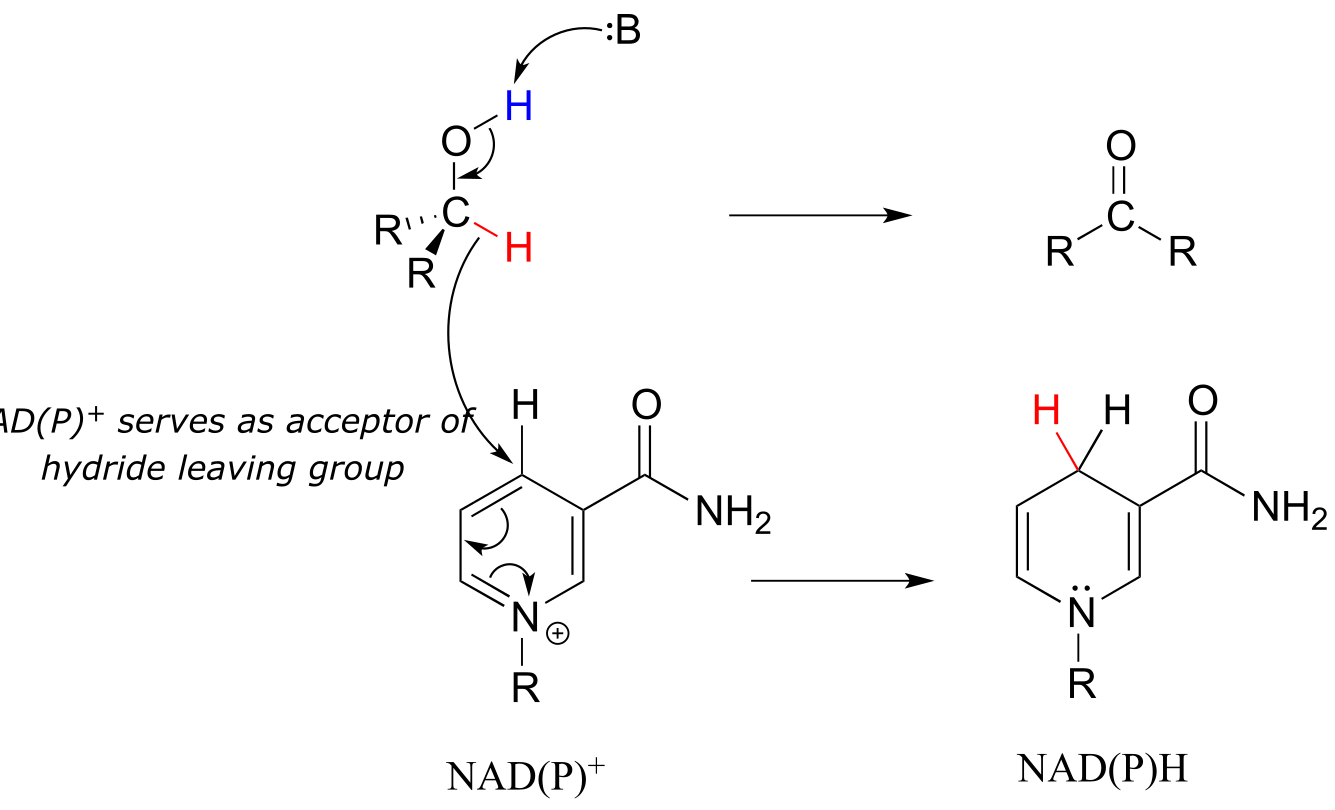
fig 19 fig 19a
An enzymatic base positioned above the carbonyl removes a proton, and the electrons in the O-H bond shift down and push out the hydride, which shifts over to carbon #4 of NAD+. Note that the same process with a primary alcohol would yield an aldehyde instead of a ketone.
Exercise 15.4: Draw general mechanisms for:
a) hydrogenation of an imine
b) dehydrogenation of an amine
Exercise 15.5: We just saw that when the nucleophile in a nucleophilic carbonyl addition step is a hydride ion from NADH, the result is a ketone/aldehyde hydrogenation reaction. As a review: what kind of reaction step results when the nucleophile in this process is not a hydride ion but a) an alcohol, or b) an enolate carbon?
The nicotinamide coenzymes also serve as hydride donors/acceptors in the redox reactions interconverting carboxylic acid derivatives and aldehydes. Notice that these reactions can be thought of as nucleophilic acyl substitution reactions (chapter 11) in which the nucleophile or leaving group is a hydride ion.
NAD(P)H-dependent hydrogenation (reduction) of a thioester to an aldehyde:
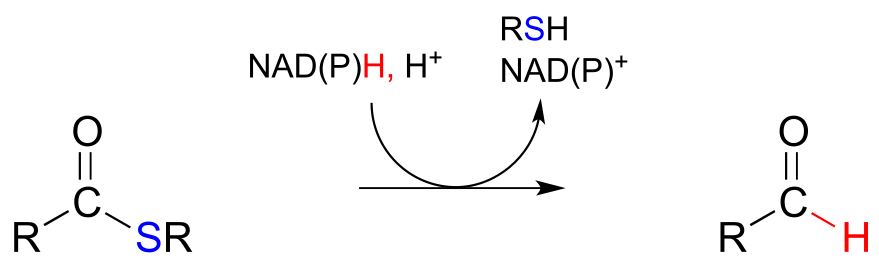
Mechanism:
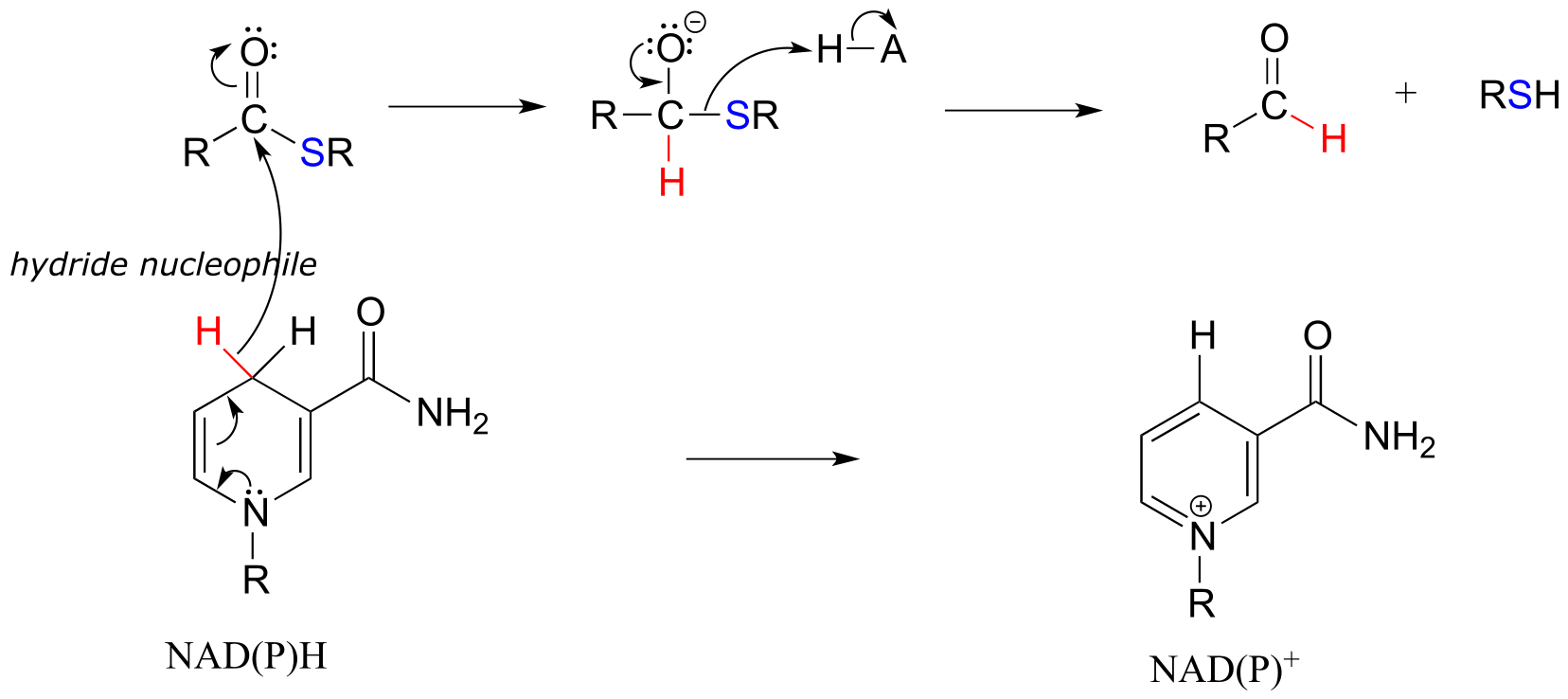
fig 20 fig 20a
NAD(P)+-dependent dehydrogenation (oxidation) of an aldehyde to a thioester:
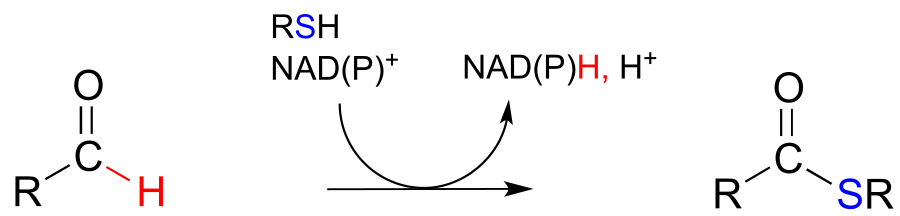
Mechanism:
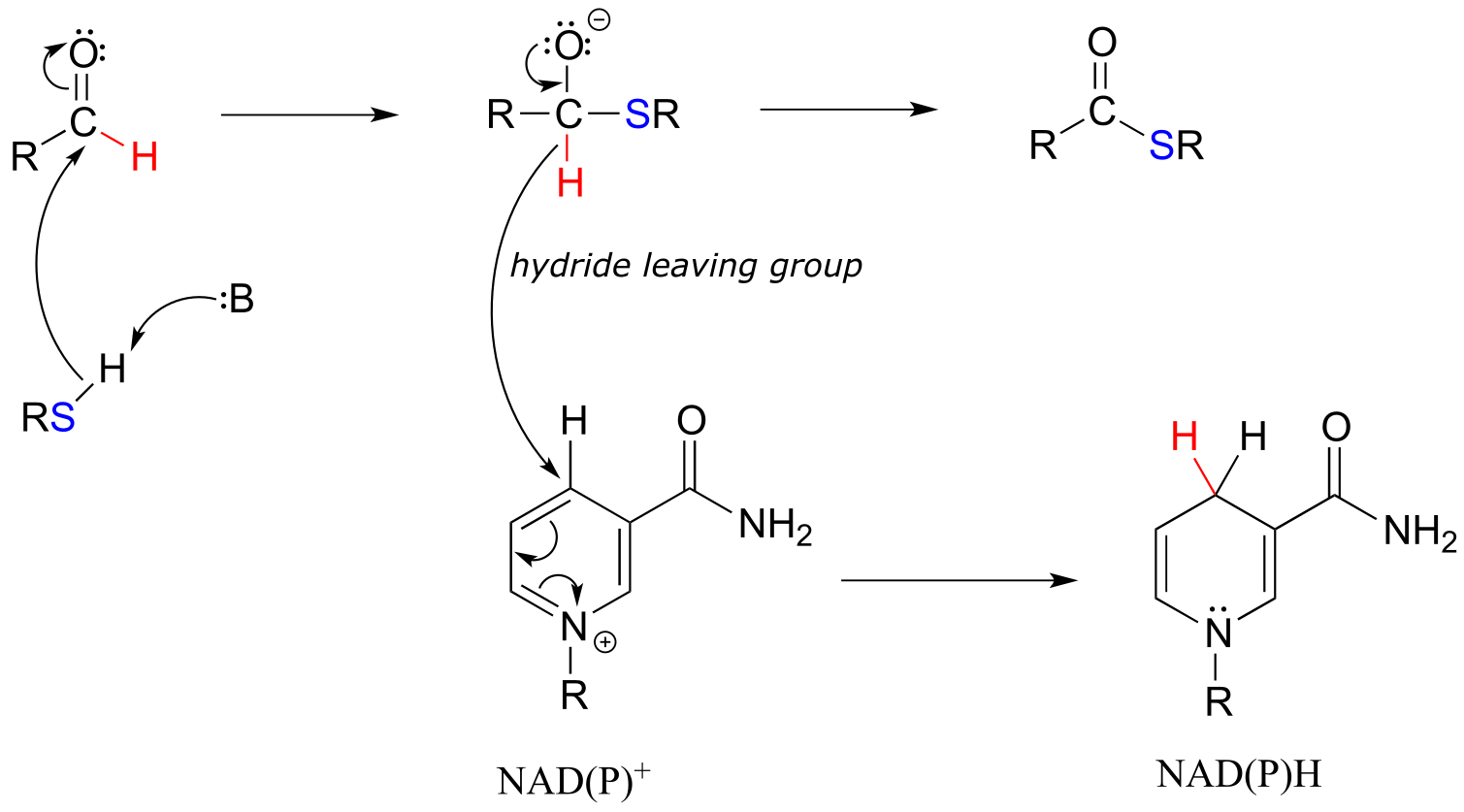
fig 21 fig 21a
To simplify figures, hydrogenation and dehydrogenation reactions are often drawn with the role of the coenzyme abbreviated:
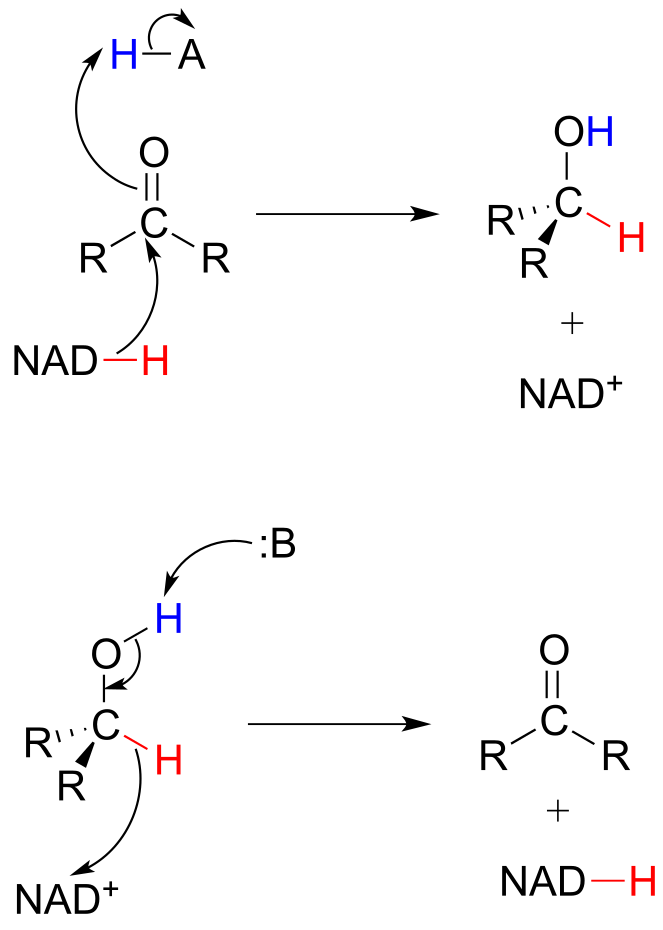
fig 22
However, it is very important to make sure that you can remember and draw out the full mechanism, including the role of the coenzyme, in these types of reactions.
Caution! A very common error made by students learning how to draw biochemical redox mechanisms is to incorrectly show nicotinamide coenzymes acting as acids or bases. Remember: NADH and NADPH are hydride donors, NOT proton donors. NAD+ and NADP+ are hydride acceptors, NOT proton acceptors.
15.3C: Stereochemistry of ketone hydrogenation#
It should not surprise you that the stereochemical outcomes of enzymatic hydrogenation / dehydrogenation steps are very specific. Consider the hydrogenation of an asymmetric ketone: In the hydrogenation direction, attack by the hydride can occur from either the re or the si face of an asymmetric ketone (see section 10.1C), leading specifically to the S or R alcohol.
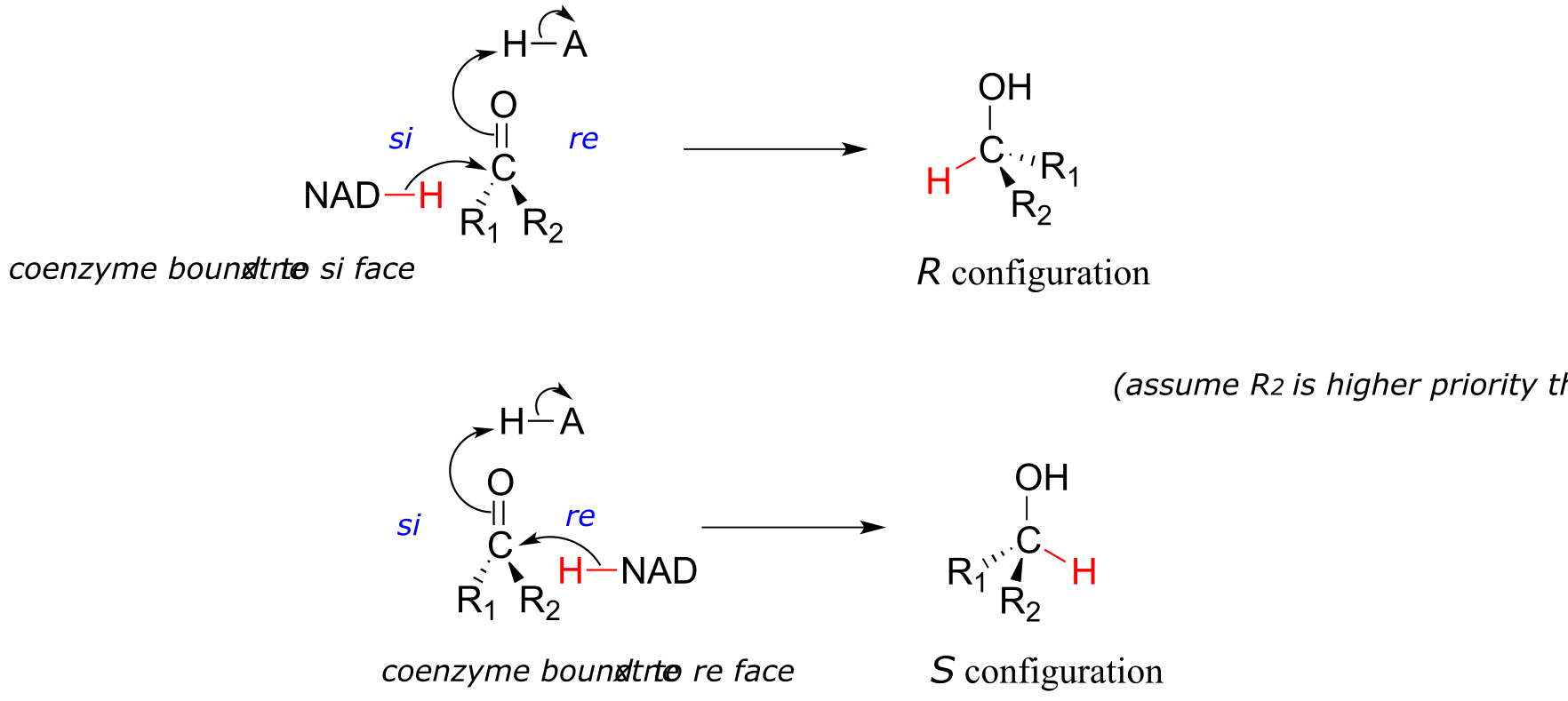
fig 23
The stereochemical configuration of the product depends on which side of the ketone substrate the NAD(P)H coenzyme is bound in the active site. Any given enzyme will catalyze its reaction with one of these two stereochemical outcomes, not both.
Stereochemical considerations apply in the dehydrogenase direction as well: in general, enzymes specifically catalyze the oxidation of either an R or S alcohol, but not both.
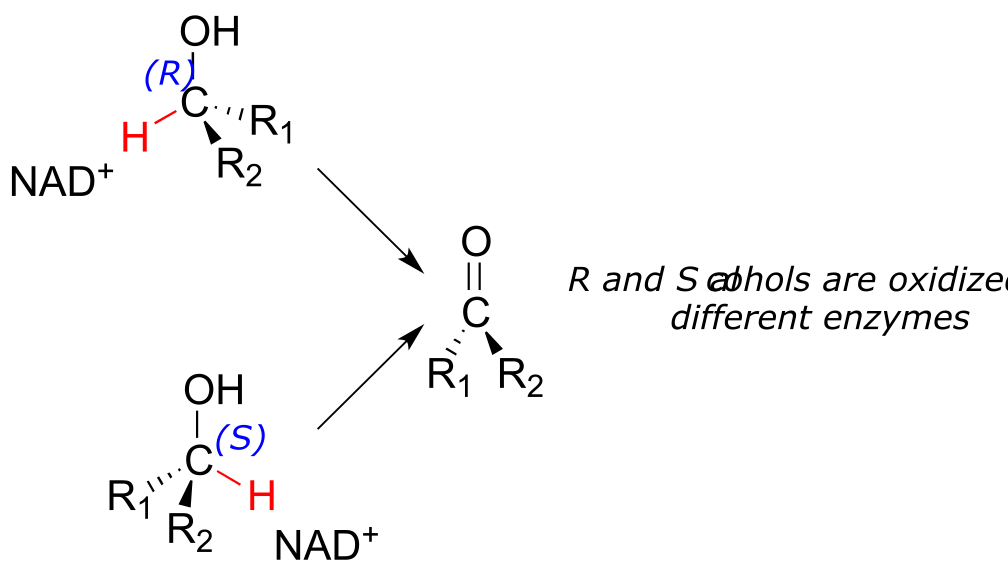
fig 24
Exercise 15.6: During an intense workout, lactic acid forms in muscle tissue as the result of enzymatic reduction of a ketone group in the precursor molecule (EC 1.1.1.27). It is the lactate that you can blame for the sore muscles you feel the day after a workout.
fig 25
a) Draw the structure of the starting ketone in this reaction.
b) Which face of the ketone is the coenzyme positioned next to in the active site of the enzyme?
15.3D: Examples of biochemical carbonyl/imine hydrogenation
Now that we have covered the basics, let’s look at some real examples of hydrogenation and dehydrogenation reactions.
Glycerol phosphate dehydrogenase (EC 1.1.1.8) catalyzes one of the final chemical steps in the breakdown of fat molecules. The enzyme specifically oxidizes (R)-glycerol phosphate to dihydroxyacetone phosphate. (S)-glycerol phosphate is not a substrate for this enzyme. J. Mol. Biol. 2006, 357, 858 (human crystal structure)
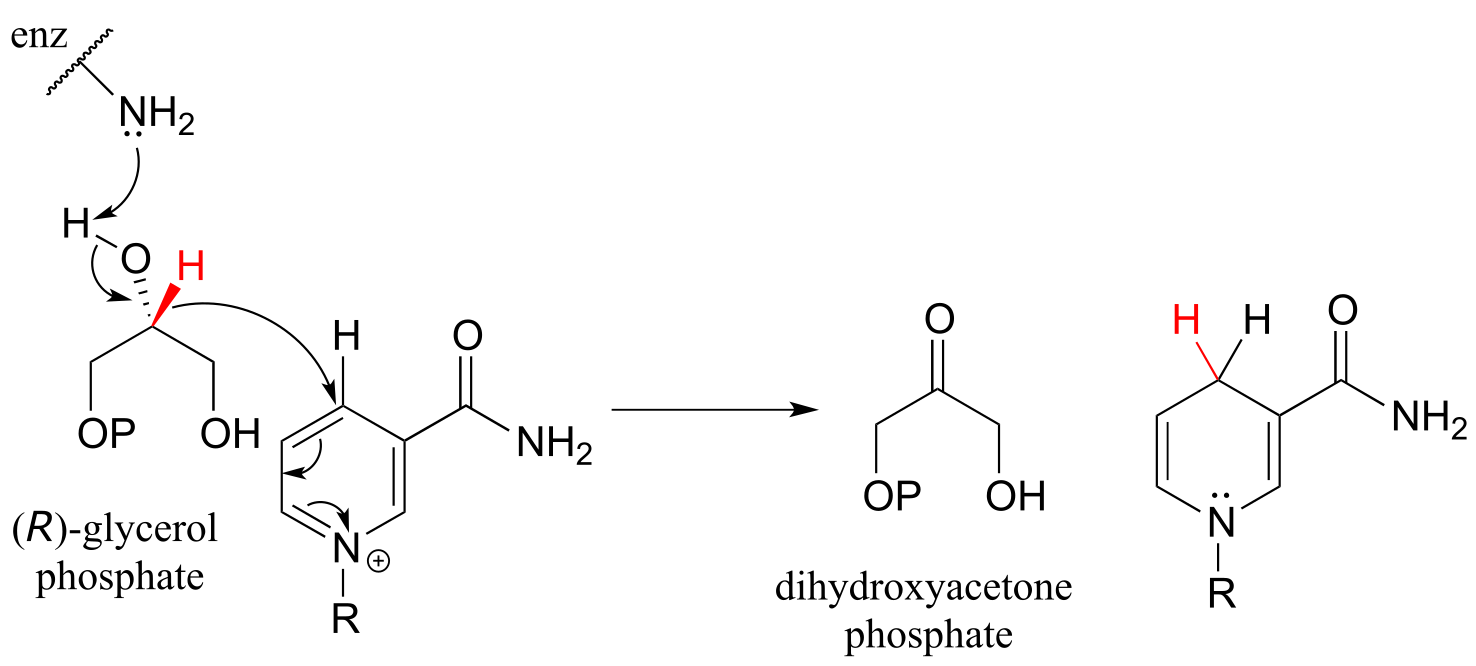
fig 26
The reverse reaction (catalyzed by the same enzyme) converts dihydroxyacetone phosphate to (R)-glycerol phosphate, which serves as a starting point for the biosynthesis of membrane lipid molecules (see section 1.3A).
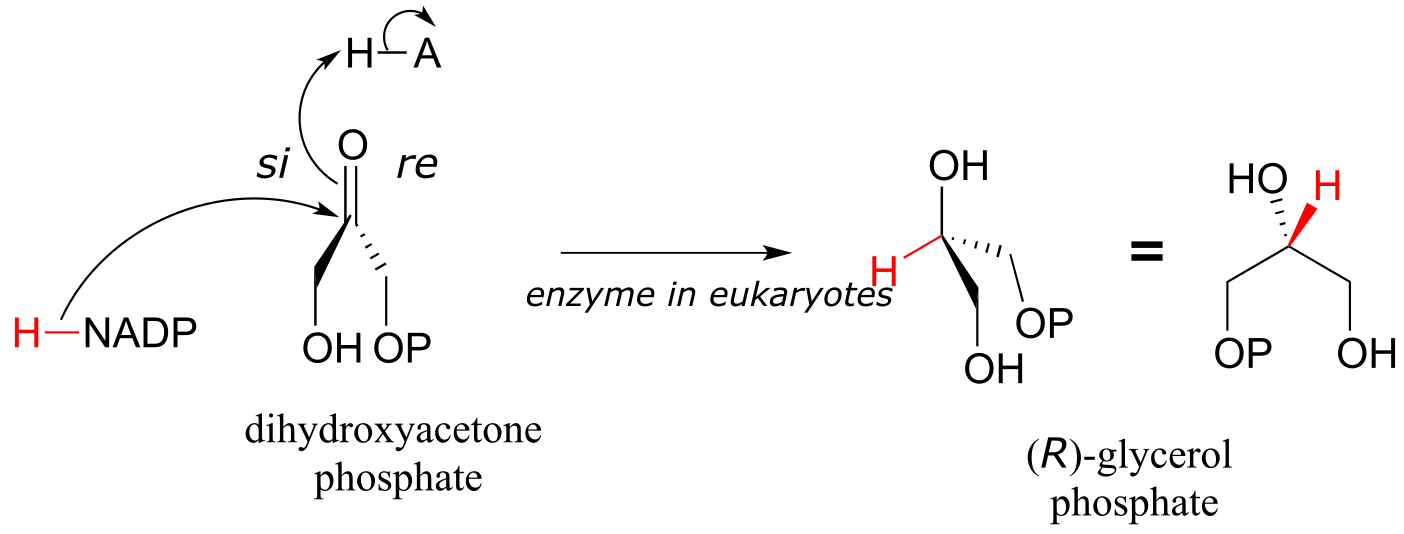
fig 26a
Exercise 15.7: X-ray crystallography experiments reveal that in the active site of glycerol phosphate dehydrogenase, a zinc cation (Zn+2) is coordinated to the oxygen atom of the carbonyl/alcohol group. How does this contribute to catalysis of the reaction?
While the cell membranes of animals, plants, and bacteria are made from lipids with the R stereochemistry exclusively, archaeal microbes (the so-called ‘third kingdom of life) are distinguished in part by the S stereochemistry of their membranes.
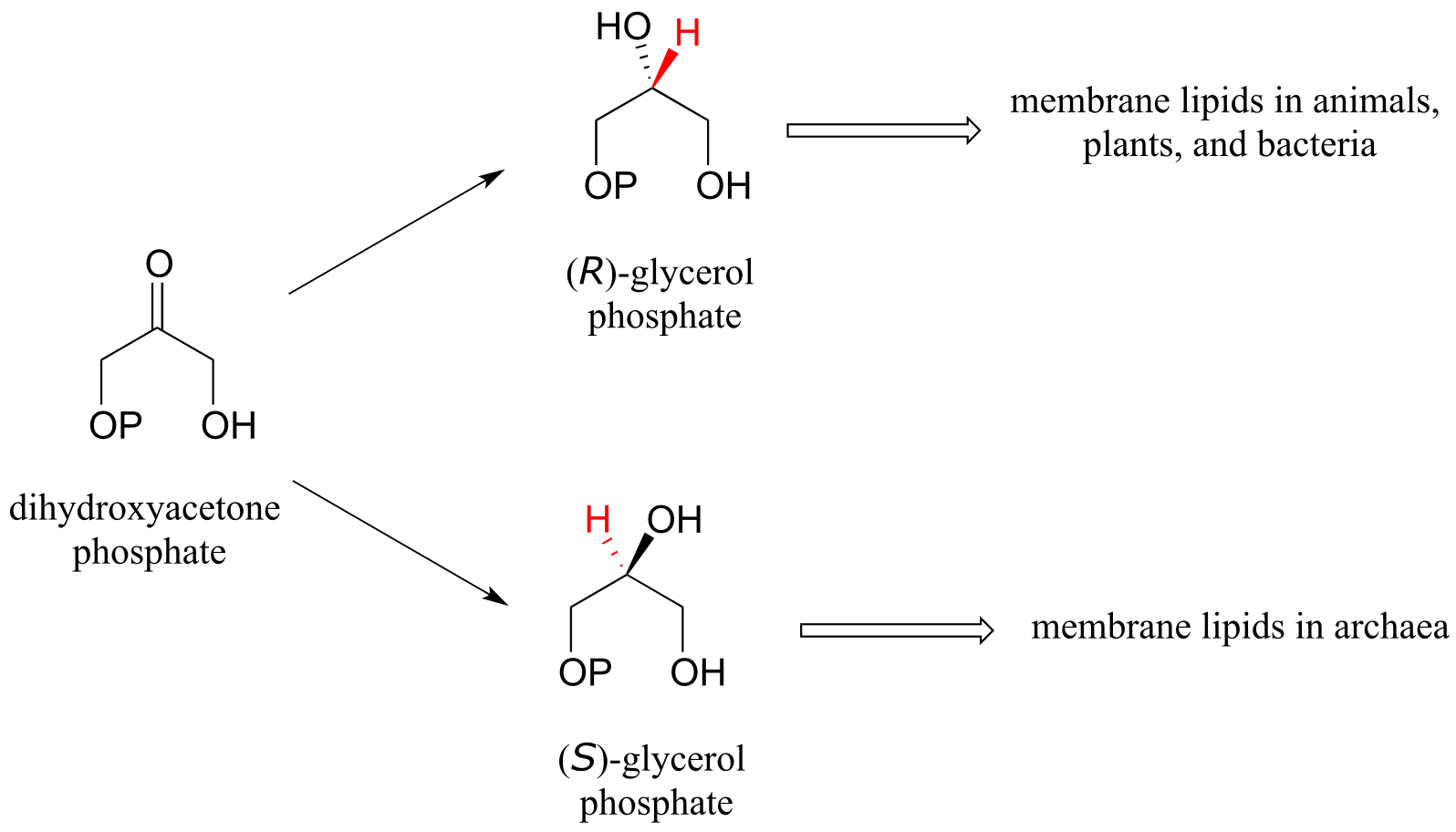
fig 27
Archaea have an enzyme that catalyzes hydrogenation of dihydroxyacetone with the opposite stereochemistry compared to the analogous enzyme in bacteria and eukaryotes. This archaeal enzyme was identified and isolated in 1997.

fig 28
In a reaction that is relevant to people who enjoy the occasional ‘adult beverage’, an NADH-dependent enzyme (EC 1.1.1.1) in brewer’s yeast produces ethanol by reducing acetaldehyde. This is the final step in the process by which yeast ferment glucose to ethanol.
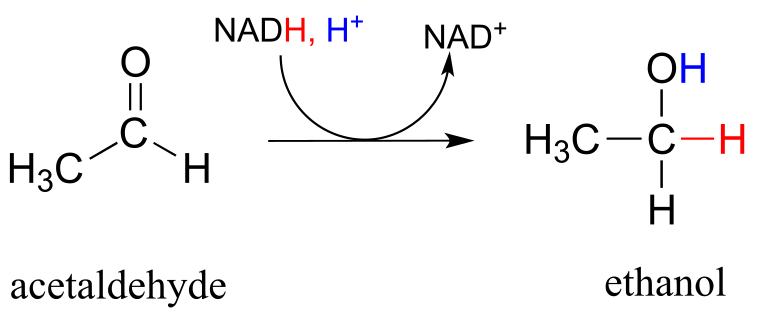
fig 29
The reaction below, which is the final step in the biosynthesis of proline (EC 1.5.1.2), is an example of an enzymatic reduction of an imine to an amine. J. Mol. Biol. 2005, 354, 91.
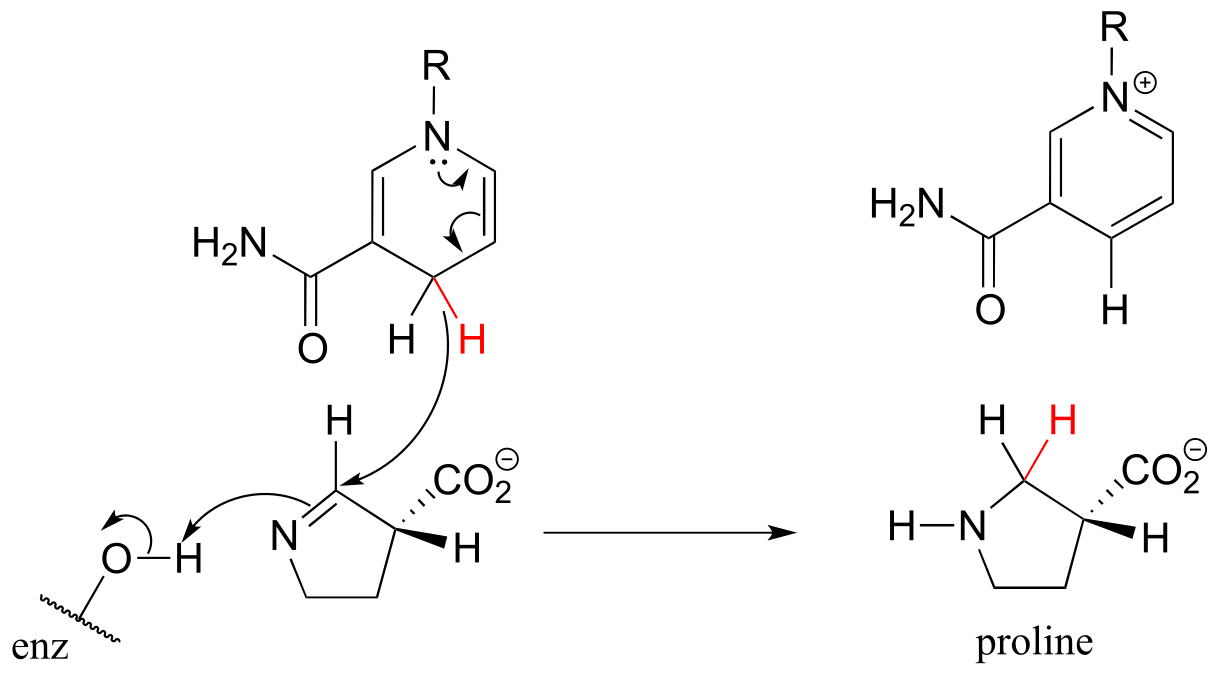
fig 30
This step in the breakdown of the amino acids glutamate (EC 1.4.1.2) provides an example of the oxidation of an amine to an imine: Structure 1999, 7, 769.
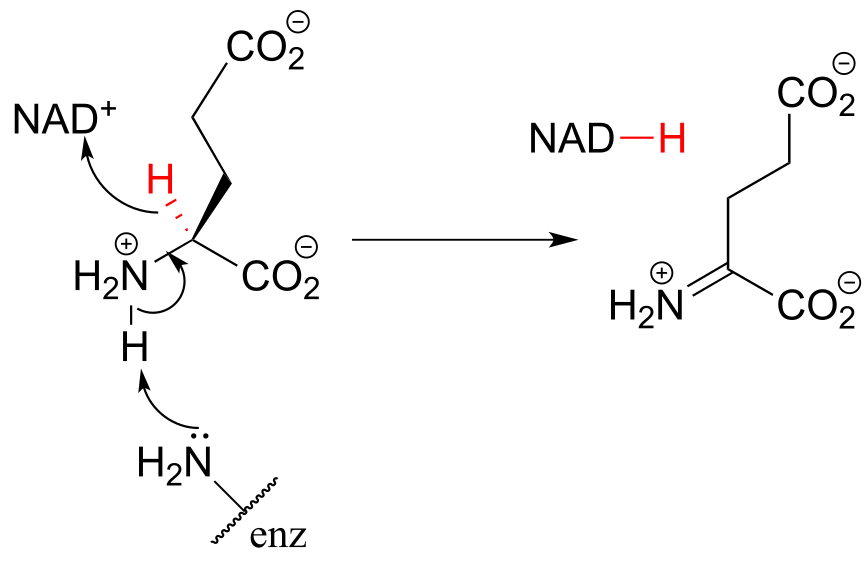
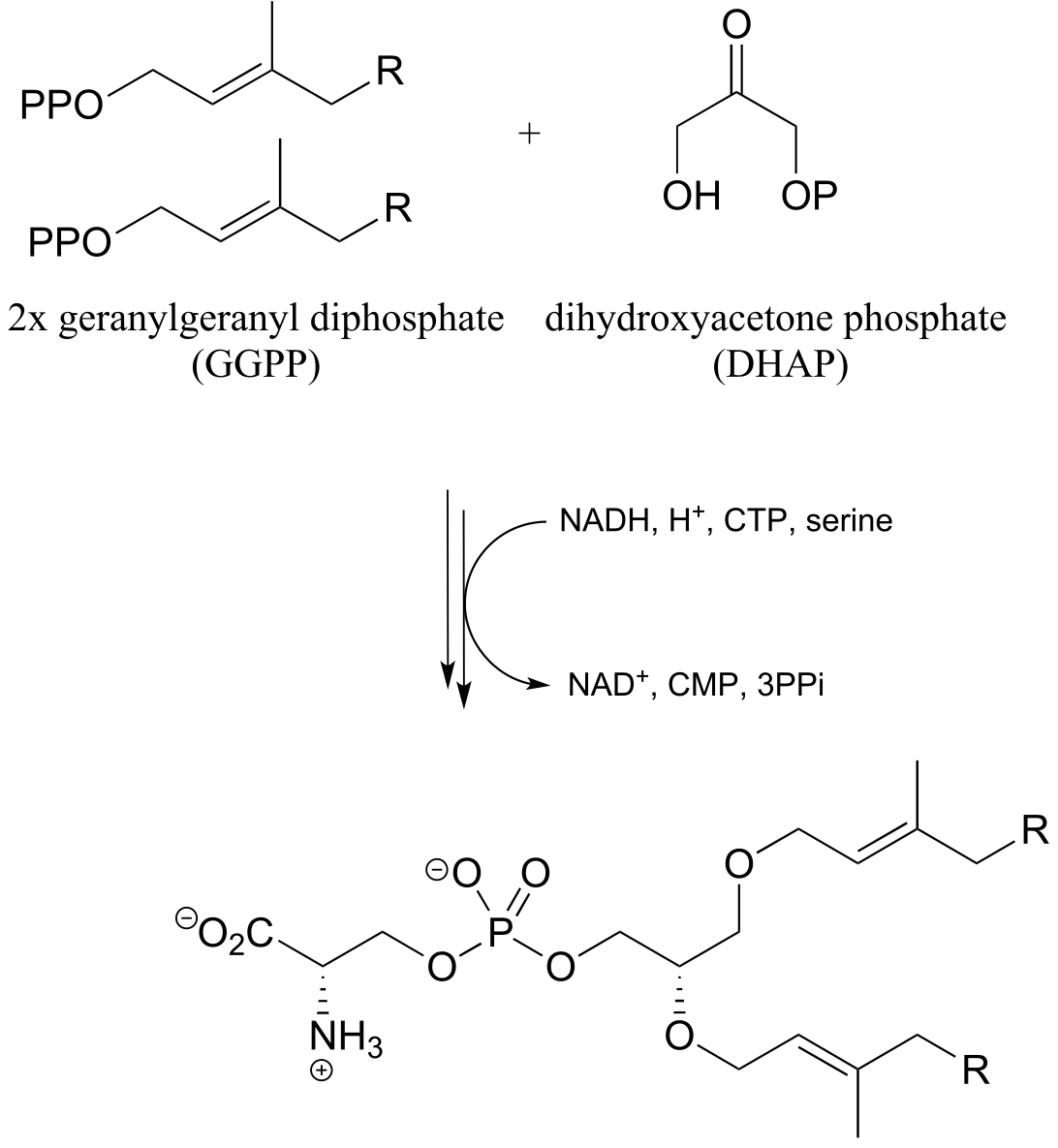
The ‘double reduction’ reaction below (EC 1.1.1.34) is part of the isoprenoid biosynthetic pathway, which eventually leads to cholesterol in humans.
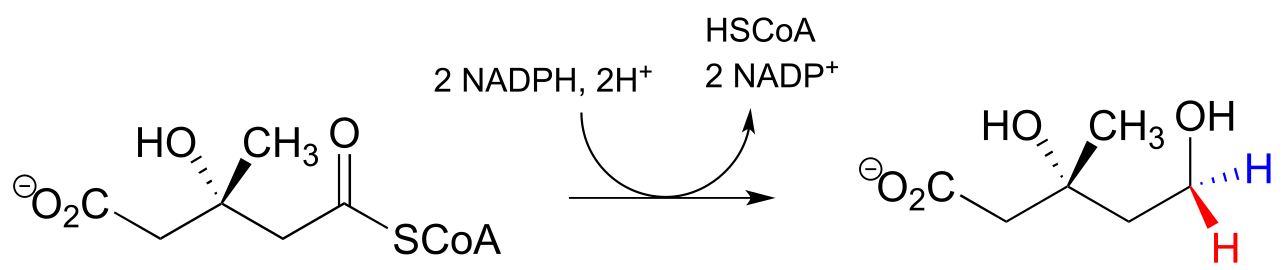
fig 32a
In this reaction, a thioester is first reduced to an aldehyde in steps 1a and 1b:
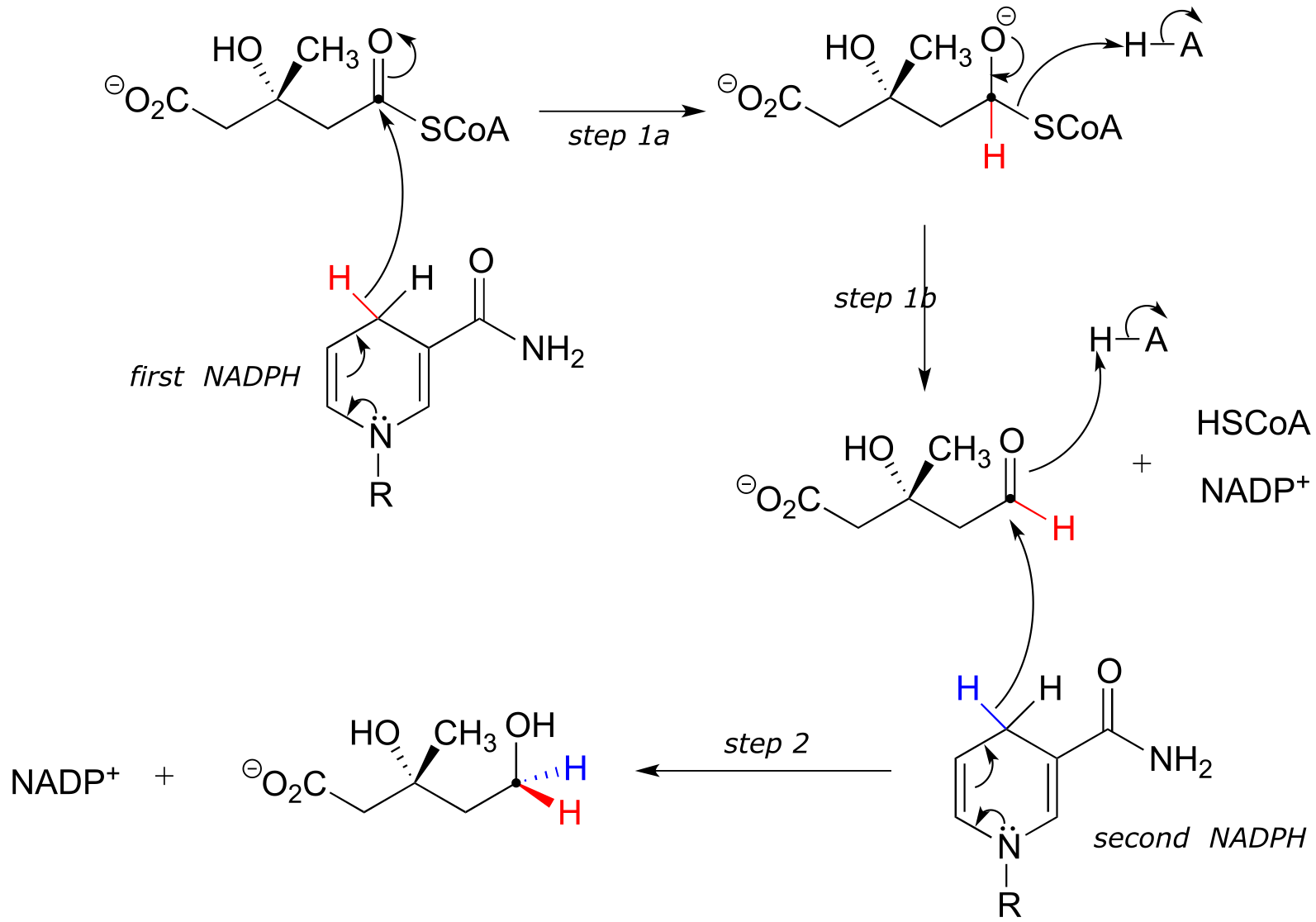
Then in step 2, the aldehyde is in turn reduced by the same enzyme (and a second NADPH that enters the active site) to a primary alcohol. This enzyme is inhibited by atorvastatin and other members of the statin family of cholesterol-lowering drugs. Atorvastatin, marketed under the trade name Lipitor by Pfizer, is one of the all-time best-selling prescription medications.
Recall from chapter 11 that carboxylates are not reactive in acyl substitution steps, so it follows that they cannot be directly reduced to aldehydes by an enzyme in the same way that thioesters can. However, a carboxylate can be converted to its ‘activated’ acyl phosphate form (section 11.4), which can then be hydrogenated. An example of this is found in a two-reaction sequence found in amino acid metabolism (EC 2.7.2.11; EC1.2.1.41).

fig 33
Glyceraldehyde-3-phosphate dehydrogenase (EC 1.2.1.12) , a key enzyme in the glycolysis pathway, provides an example of the oxidation of an aldehyde to a thioester, in this case a thioester linkage between the substrate and a cysteine residue in the enzyme’s active site. In the second phase of the reaction, the thioester intermediate is hydrolyzed to free the carboxylate product.
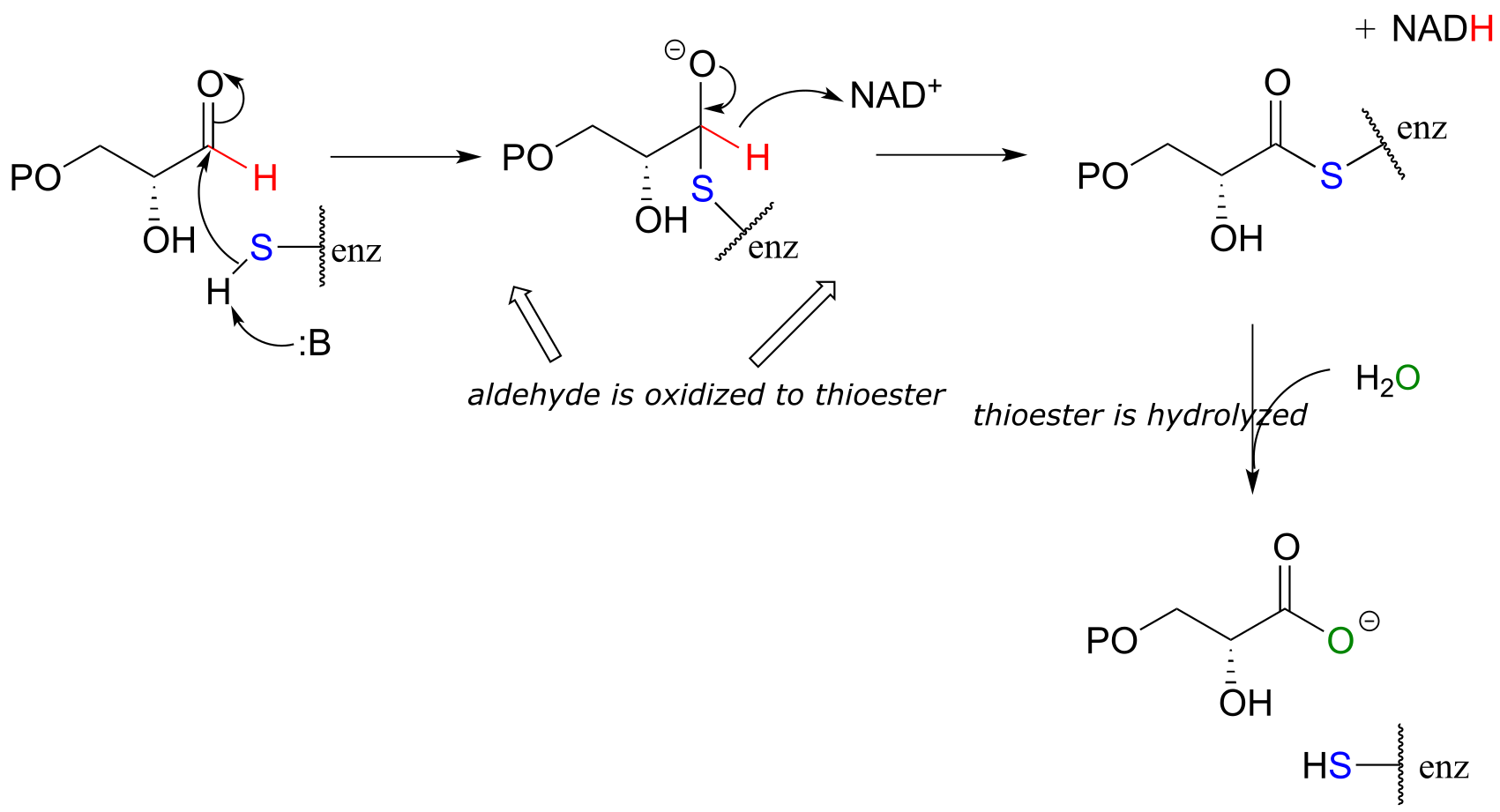
fig 34
Exercise 15.8: Below is the final step in the biosynthesis of the amino acid histidine (EC 1.1.1.23). Fill in the species that are indicated with question marks. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 1859
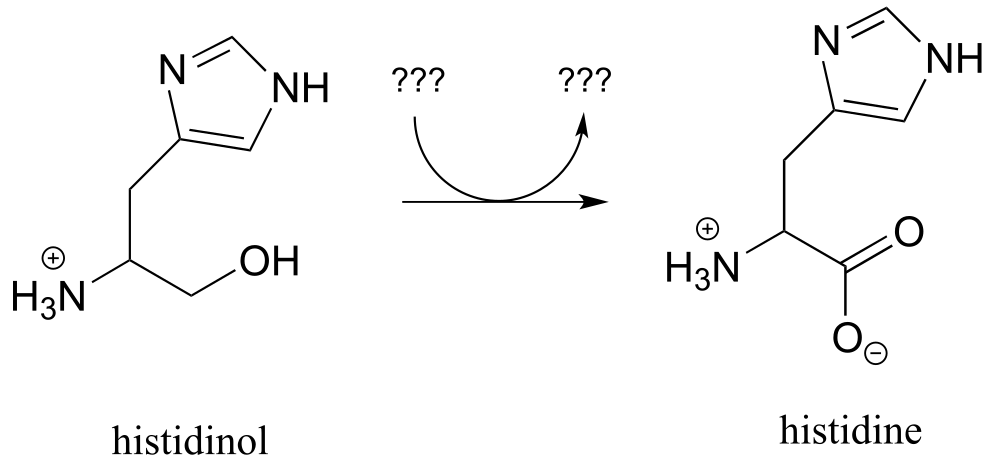
fig 35
Exercise 15.9: Draw a likely mechanism for the conversion of glucose to sorbitol, a process that occurs in the liver. Do not abbreviate the nicotinamide ring structure.
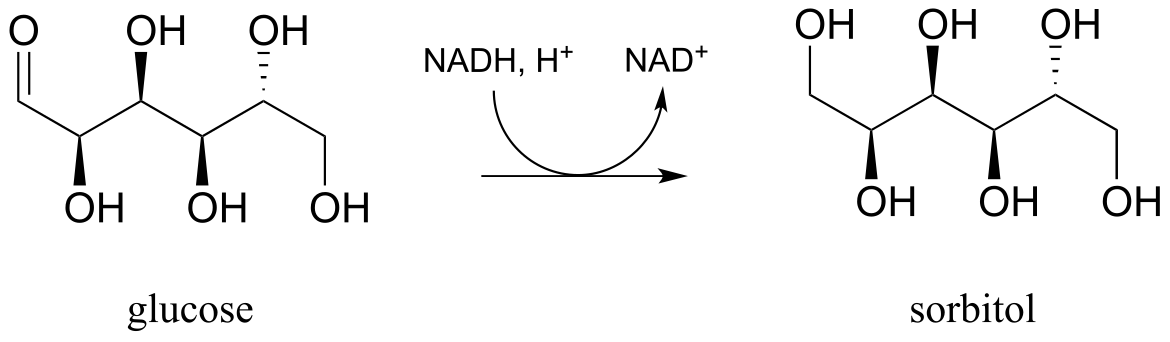
fig 35a
15.3E: Reduction of ketones and aldehydes in the laboratory#
Although our focus in this book is biological organic reactions, it is interesting to note that synthetic organic chemists frequently perform hydrogenation reactions in the lab that are similar in many respects to the NAD(P)H-dependent reactions that we have just finished studying. A reagent called sodium borohydride (NaBH4) is very commonly used, often in methanol solvent, to reduce ketones and aldehydes to alcohols. The reagent is essentially a laboratory equivalent of NADH (or NADPH): it serves as a source of nucleophilic hydride ions. Sodium borohydride is a selective reagent in the sense that it will reduce ketones and aldehydes but not carboxylic acid derivatives such as esters (recall from section 11.2 and 11.3 that the carbonyl carbons of carboxylic acid derivatives are less potent electrophiles than the carbonyl carbons of ketones and aldehydes). Unlike the enzymatic hydrogenation reactions we saw earlier, the reduction of asymmetric ketones with sodium borohydride usually results in a 50:50 racemic mixture of the R and S enantiomers of the alcohol product.
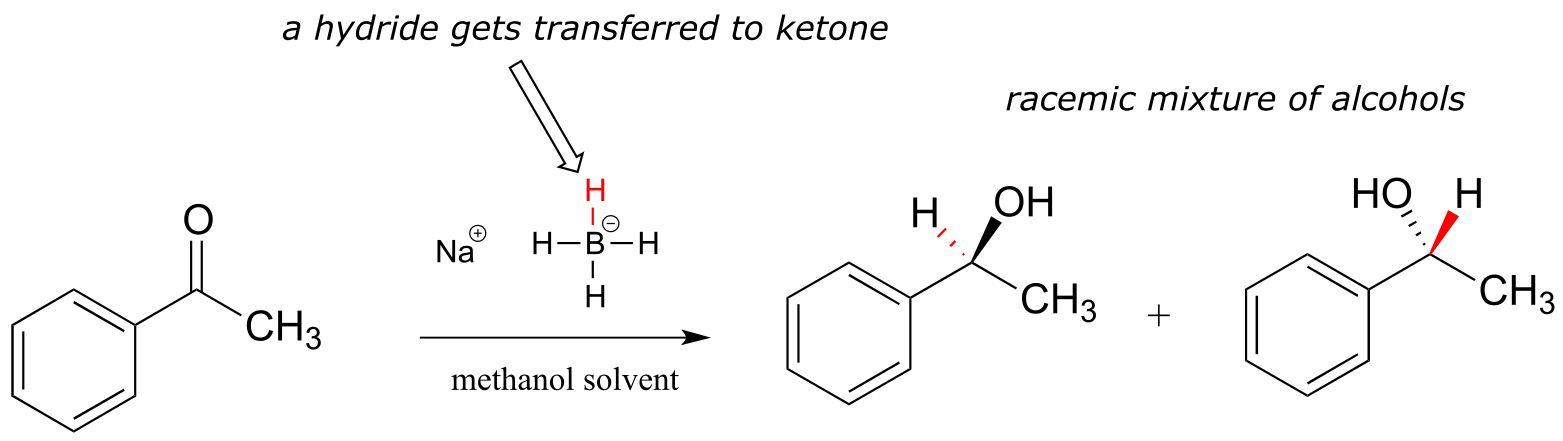
fig 36
Synthetic organic chemists have at their disposal a wide range of other reducing and oxidizing reagents with varying specificities and properties, many of which you will learn about if you take a course in laboratory synthesis.
Exercise 15.10: Camphor can be easily reduced by sodium borohydride. However, the mixture of stereoisomeric alcohols that results is not 50:50.
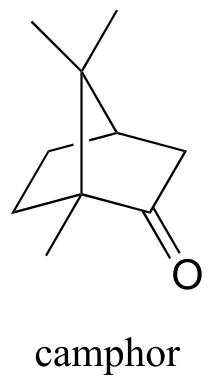
fig 37
a) Draw the two stereoisomers of the alcohol products of this reaction, and explain why they are not formed in a 50:50 ratio.
b) Which analytical technique - 1H-NMR, IR, UV, or MS - could best be used to determine the ratio of the stereoisomers in the product mix? Describe how this analysis could be accomplished.
Section 15.4: Hydrogenation of alkenes and dehydrogenation of alkanes#
We turn next to reactions in which a hydrogen molecule is added to the double bond of an alkene, forming an alkane - and the reverse, in which H2 is eliminated from an alkane to form an alkene. Many biochemical reactions of this type involve α,β-unsaturated thioesters.
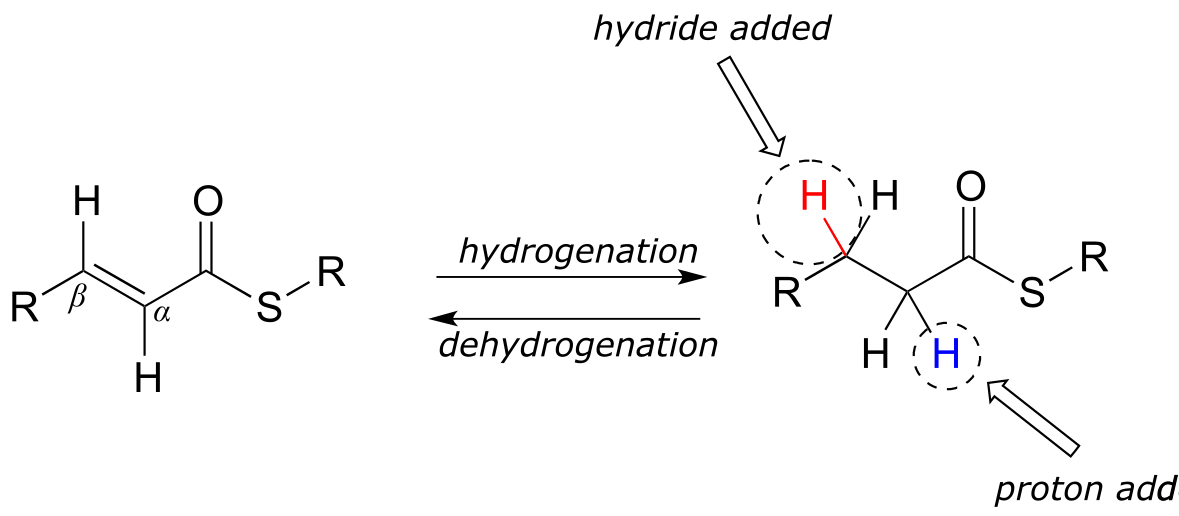
fig 38
15.4A: Alkene hydrogenation#
In the cell, alkene hydrogenation most often occurs at the α and β position relative to a carbonyl. This type of alkene hydrogenation is essentially a conjugate addition (section 11.4) of hydrogen, with a hydride ion (often from NAD(P)H) acting as the nucleophile in the first step.
NAD(P)H-dependent hydrogenation (reduction) of an α,β-conjugated alkene:
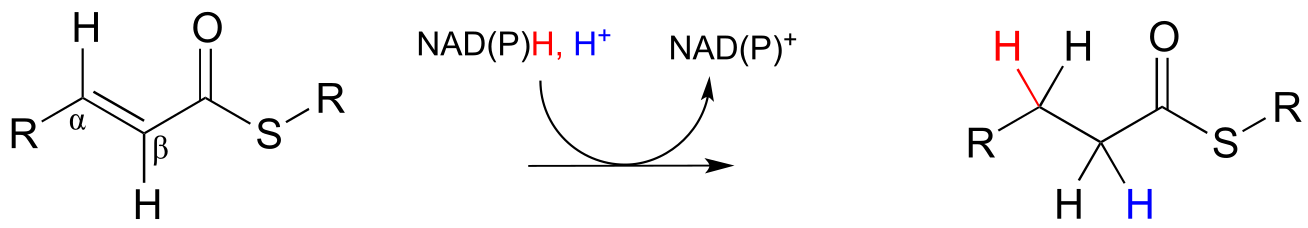
Mechanism:
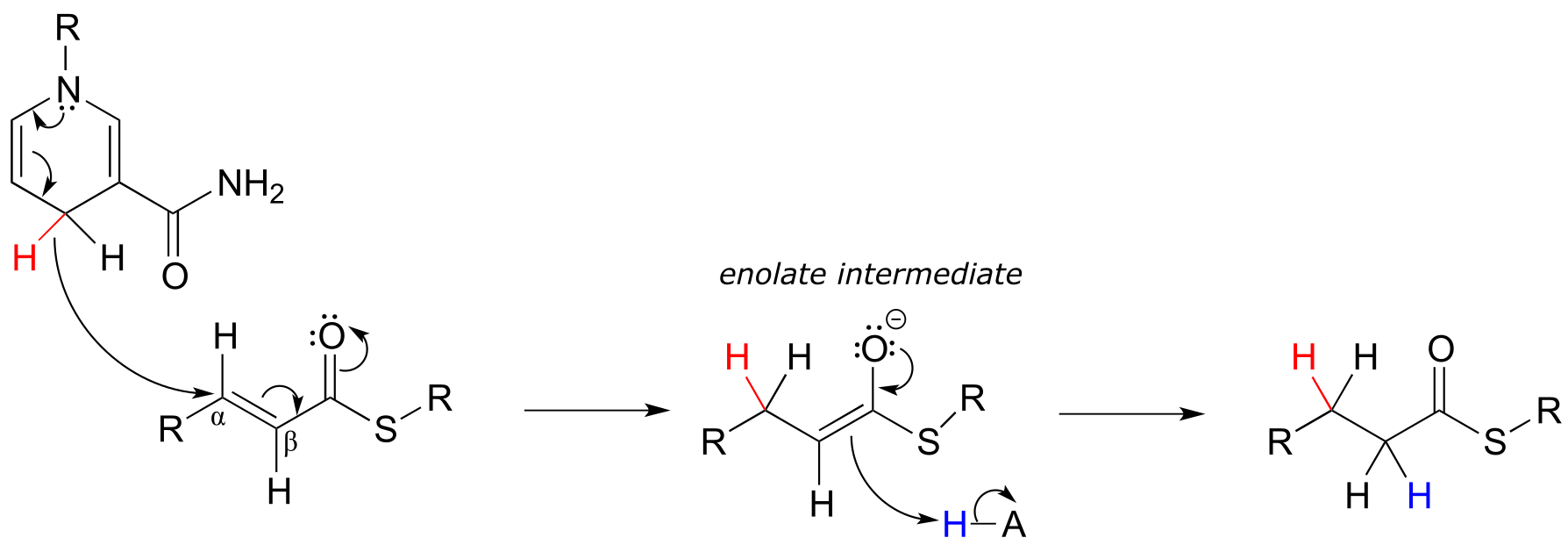
fig 39 fig 39a
As part of the fatty acid synthesis pathway, a double bond between the α and β carbons of a fatty acid is reduced to a single bond by hydrogenation (EC 1.3.1.10). The fatty acid is attached to an acyl-carrier protein via a thioester linkage (section 11.5A).
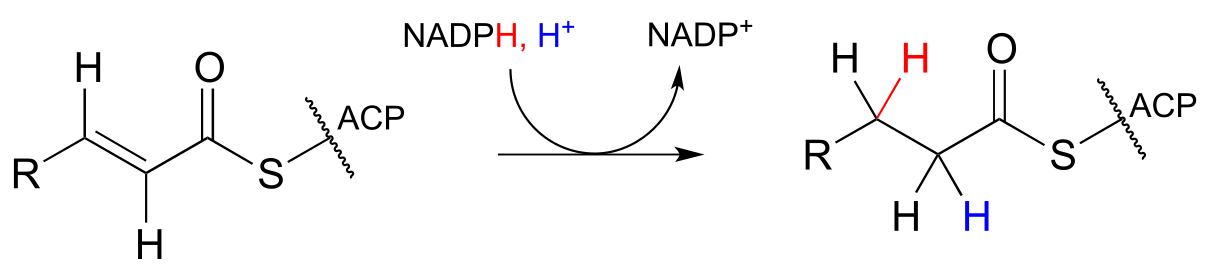
fig 40
It can be easy to forget but important to remember that there is a lot of stereospecificity inherent in biochemical reactions, including this one - even though no chiral centers are involved. First, notice that the substrate contains a trans (E) alkene. Next, let’s add some more information about prochirality:

fig40a
Notice that in this particular reaction it is specifically the pro-R hydride on NADPH that is delivered to the substrate. Also notice that both the hydride and proton are added to the re faces of the α and β carbons of the substrate, and become the pro-R and pro-S hydrogen atoms, respectively, on the product. This level of stereospecificity, you should recall from previous discussions, stems from the highly precise positioning of substrate and cofactor within the active site of the enzyme.
Other hydrogenase enzymes are known to deliver the pro-S hydride of NADH or NADPH to their substrate, and there are many examples of biochemical conjugate addition reactions in which the nucleophile and proton are added from opposite sides. Always keep in mind that stereochemistry is a key element in the amazing diversity of biological organic reactions.
15.4B: Flavin-dependent alkane dehydrogenation#
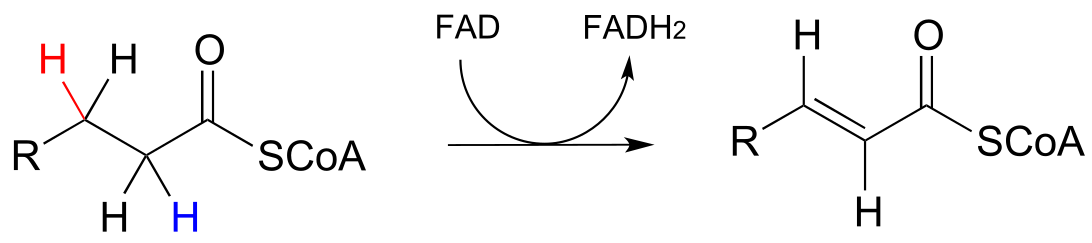
Next, let’s consider an alkane dehydrogenation reaction (EC 1.3.99.3) in the fatty acid degradation pathway. Here, a double bond is introduced between the α and β carbons, with concurrent loss of a hydride ion and a proton.
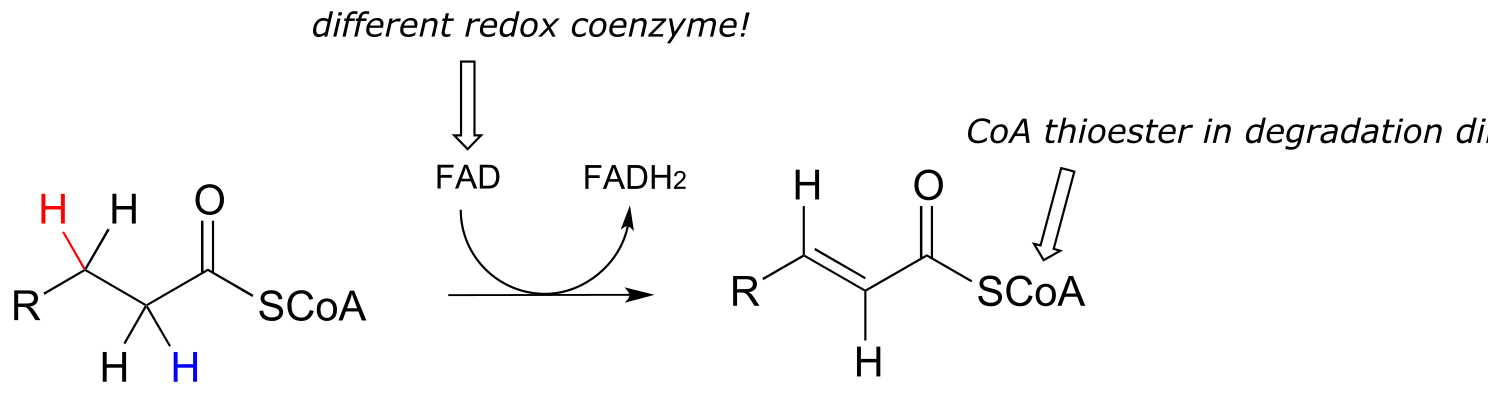
fig 41
This reaction is clearly not the reverse of the hydrogenation reaction we just saw from fatty acid biosynthesis. First of all, you should notice that the thioester linkage is to coenzyme A rather than acyl carrier protein (ACP). More importantly to this discussion, while the hydride donor in the biosynthetic hydrogenation reaction is NADPH, the relevant coenzyme in the catabolic direction is not NAD+ or NADP+ - rather, it is a flavin coenzyme.
Flavin adenine dinucleotide (FAD) is composed of three components: the three-ring flavin system, ribose phosphate, and AMP. An alternate form, which is missing the AMP component, is called flavin mononucleotide (FMN).
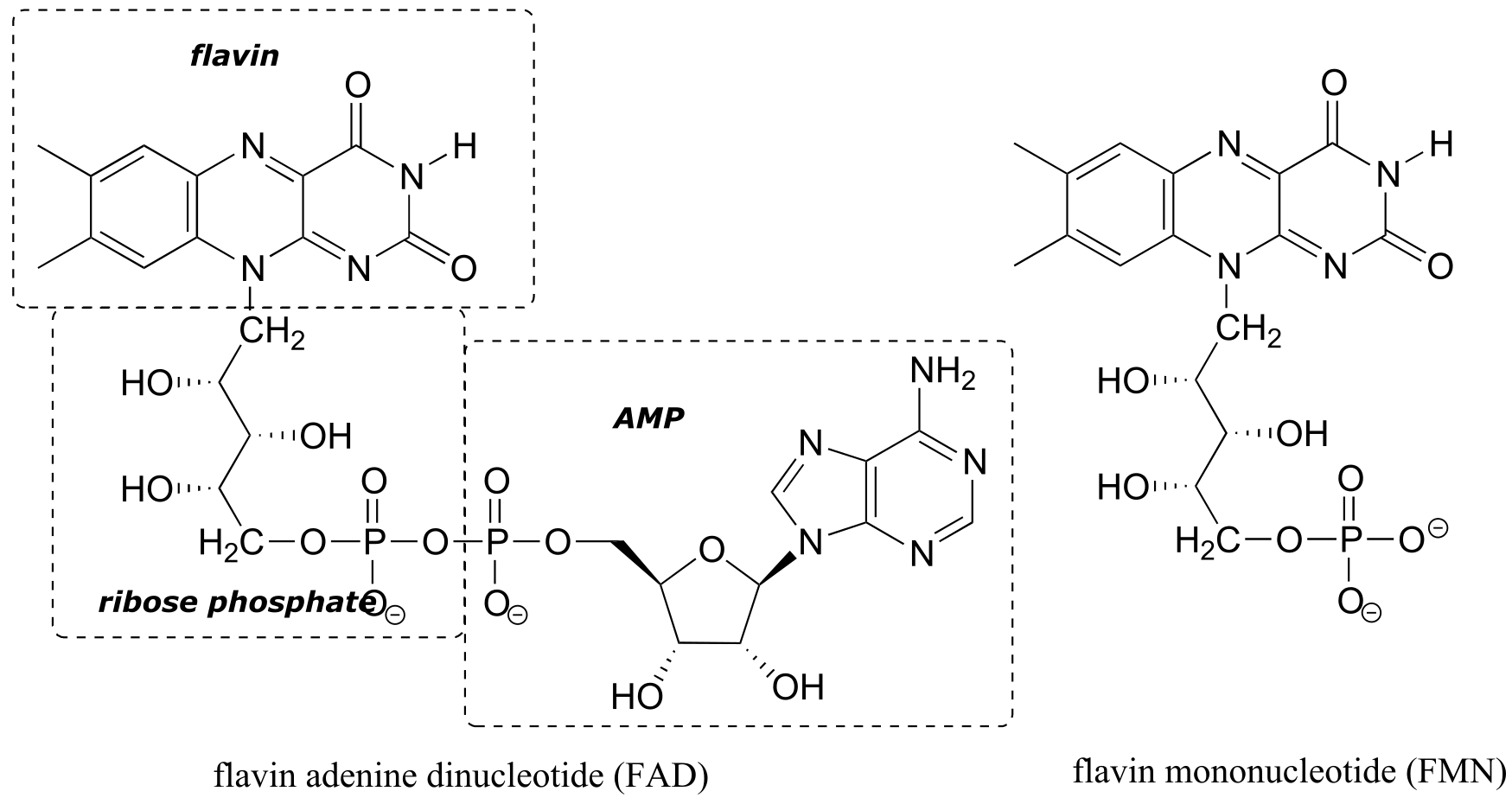
fig 42
The reactive part of the coenzyme is the flavin group, so usually the rest of the molecule is abbreviated with ‘R’.
FAD and FMN are the oxidized form of flavin. The reduced (hydrogenated) forms of these cofactors are abbreviated FADH2 and FMNH2.
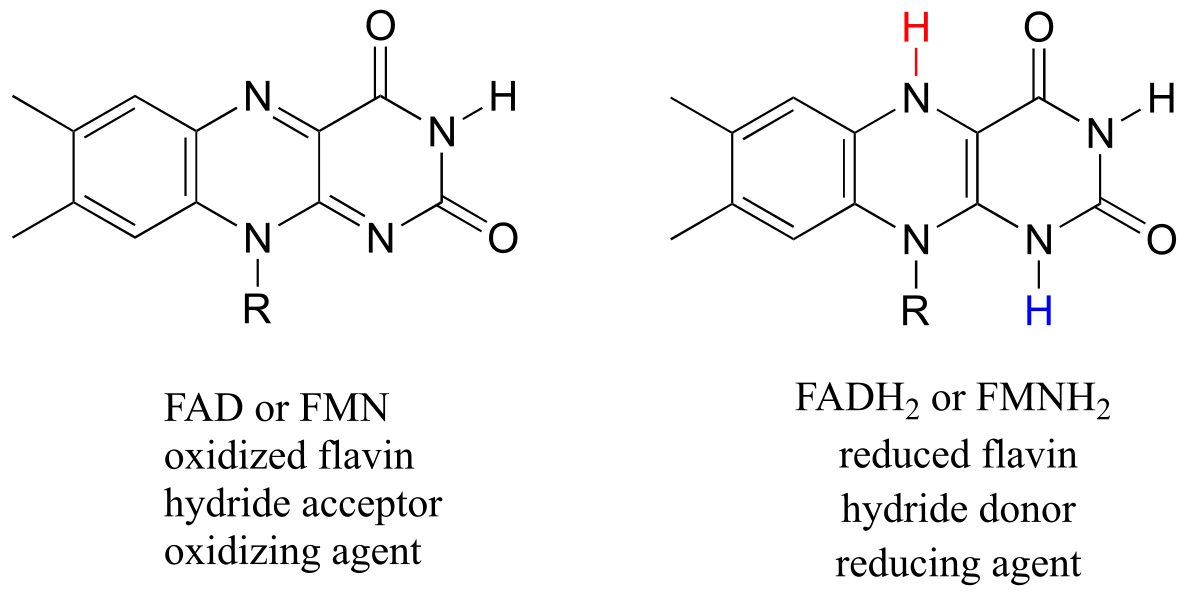
fig 43
The flavin coenzymes are synthesized in humans from riboflavin (vitamin B2), which we obtain from our diet (the structure of riboflavin is the same as that of FMN, except that riboflavin lacks the phosphate group). Notice the extended conjugated π system in the three fused rings: the flavin system absorbs light in the visible wavelengths and has a distinctive deep yellow color - it is riboflavin, and to some extent FAD and FMN, that give urine its color.
Like the nicotinamide coenzymes, flavin serves as a hydride donor or acceptor. FAD and FMN are able to accept a hydride ion (and a proton), and FADH2 and FMNH2 in turn can serve as hydride donors in hydrogenation reactions.
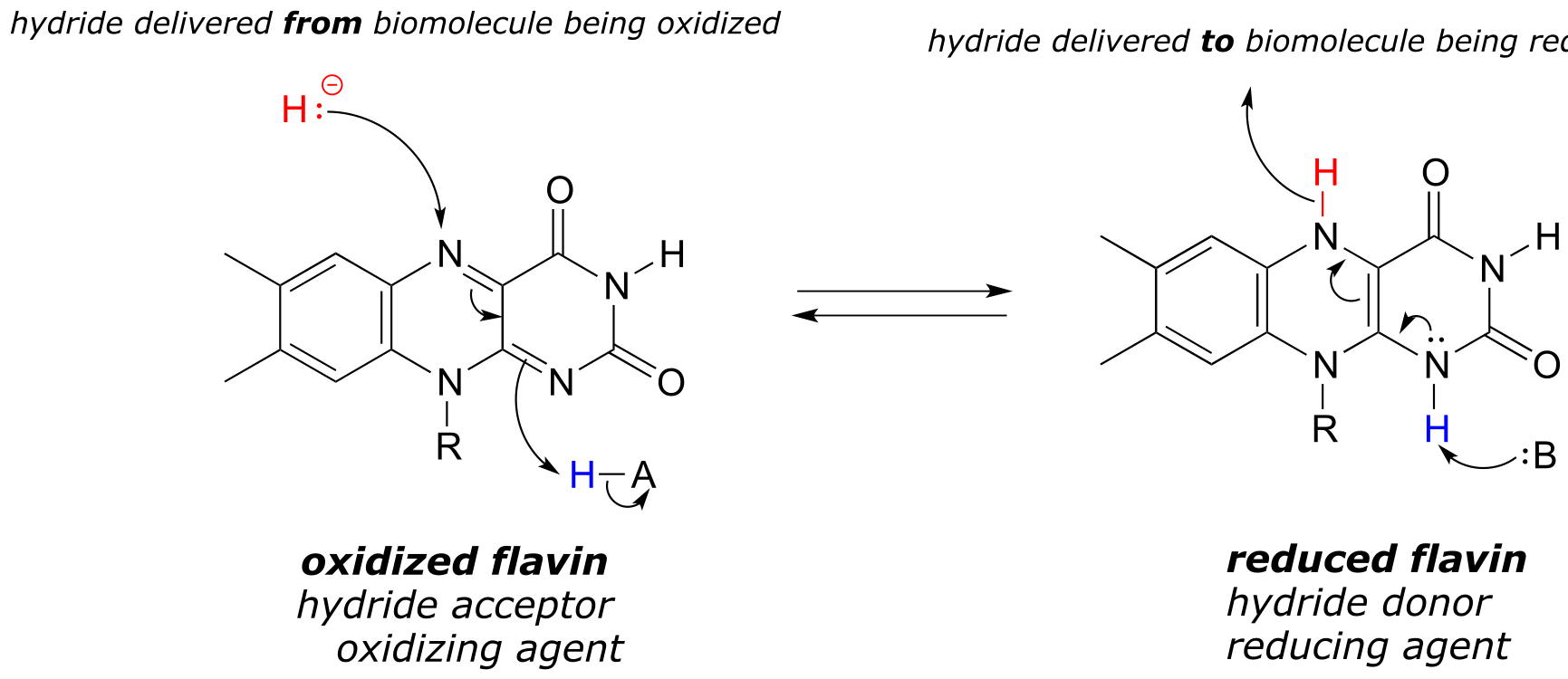
fig 44
Below is a general mechanism for the dehydration of an alkene at the α,β position - notice that it is mechanistically an E1cb elimination of H2.
Flavin-dependent α,β dehydrogenation (oxidation) of an alkane:
Mechanism:
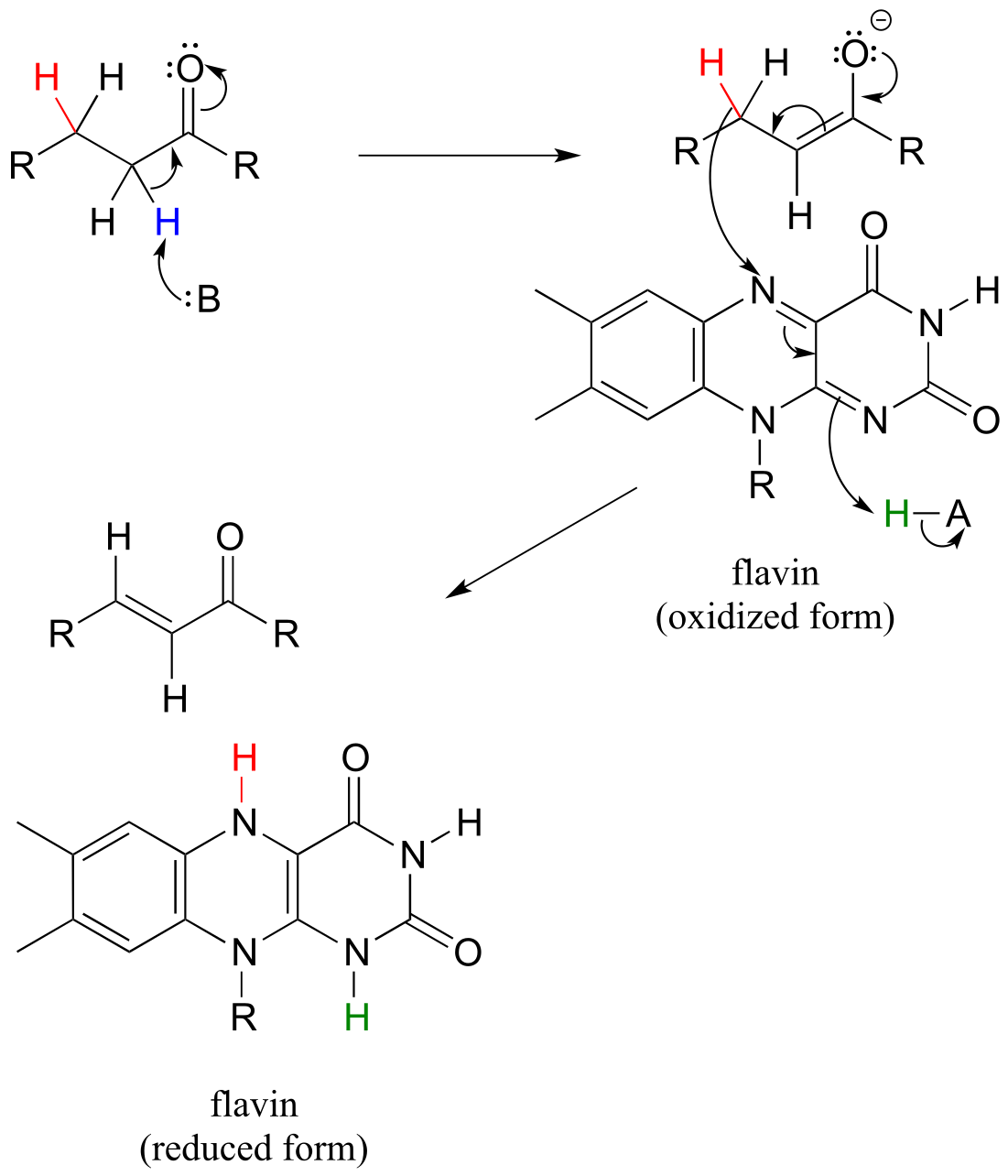
fig 45 fig 45a
In many enzymatic reactions in which FADH2 acts as the reducing agent, the reaction cycle is completed when FAD, rather than being released from the active site, is recycled back to FADH2 with the concomitant oxidation of NADH.
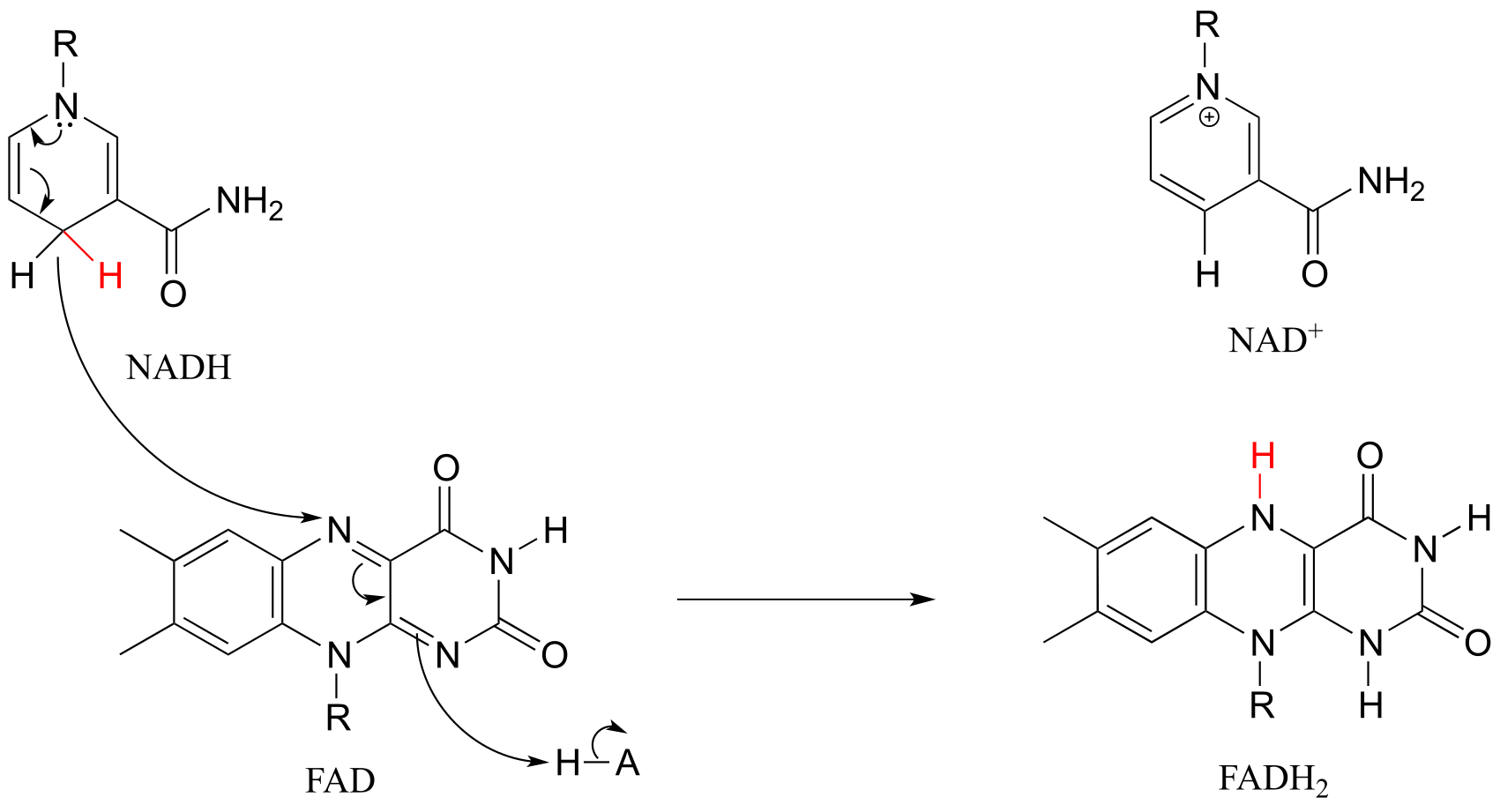
fig 51g
Hydride ion transfer with flavin or nicotinamide coenzymes is a two electron redox process. However, unlike the nicotinamide cofactors, flavins are also able to function in single electron transfer (radical) mechanisms. We will come back to this idea briefly in the chapter 16.
Exercise 15.11: Fumarate is formed in an alkane dehydrogenation reaction (EC 1.3.5.1) which is part of the citric acid cycle:
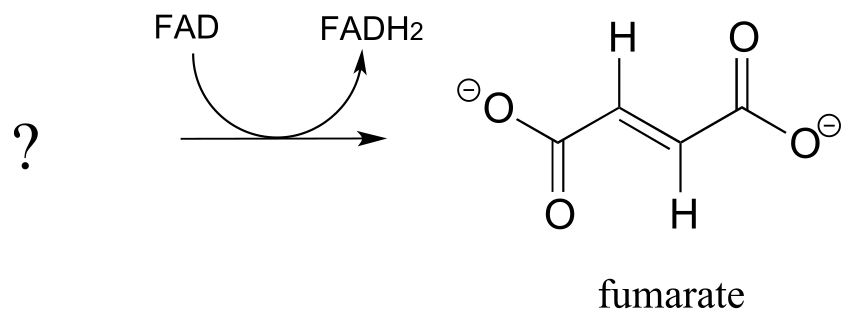
fig 46
a) Predict the structure of the starting substrate in this reaction
b) Draw the structure of the enolate intermediate
Exercise 15.12: Reduced flavin can serve as the hydride donor in some hydrogenation reactions. Degradation of the RNA base uracil begins with hydrogenation of a conjugated alkene group by a flavin-dependent hydrogenase enzyme (EC 1.3.1.2). Predict the product of this step, and draw curved arrows for the first mechanistic step.
fig 47
**
**
Section 15.5: Monitoring hydrogenation and dehydrogenation reactions by UV spectroscopy#
In order to study any enzyme-catalyzed reaction, a researcher must have available some sort of test, or assay, in order to observe and measure the reaction’s progress and measure its rate. In many cases, an assay simply involves running the reaction for a specified length of time, then isolating and quantifying the product using a separation technique such as high performance liquid chromatography (HPLC) or gas chromatography (GC). This type of assay can be extremely time-consuming, however, so it is to the researcher’s great advantage if a more convenient assay can be found.
Redox reactions in which a nicotinamide coenzyme participates as a hydride donor or acceptor are generally quite convenient to assay. In fact, the progress of these reactions can usually be observed in real time, meaning that the researcher doesn’t need to stop the reaction in order to see how far it has progressed. NADPH and NADH have distinctive n-π* UV absorbance bands centered at 340 nm, with a molar absorptivity of 6290 M-1 cm-1 (section 4.4). The oxidized coenzymes NADP+ and NAD+ do not absorb at this wavelength.
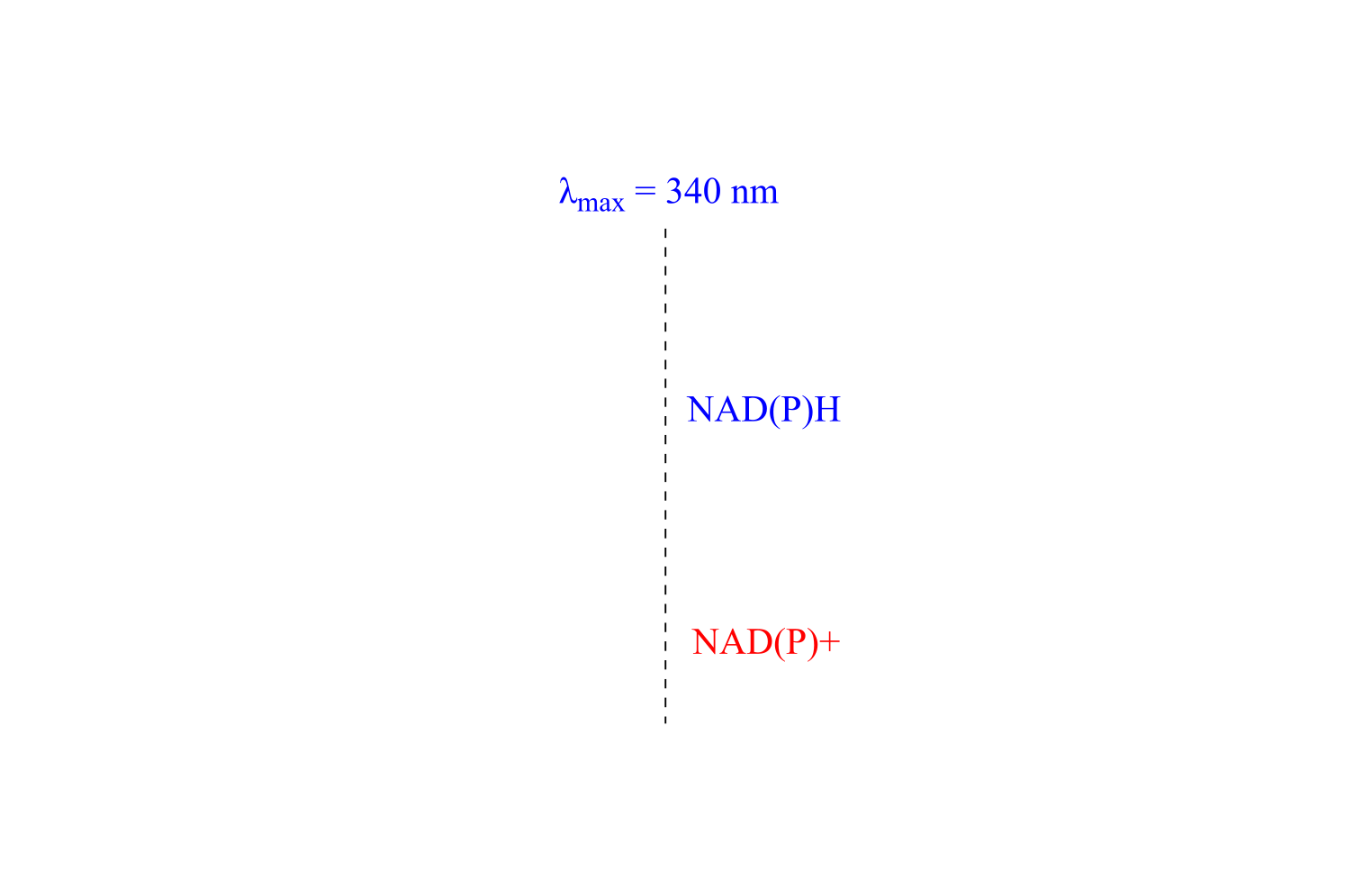
fig 48
Therefore, the course of a hydrogenation reaction, in which NAD(P)H is converted to NAD(P)+, can be observed in real time if it is run in a quartz cuvette in a UV spectrometer. By observing the decrease in absorbance at 340 nm, the researcher can calculate how much NAD(P)H has been oxidized to NAD(P)+ at any given time point, and this number is the molar equivalent of the amount of organic substrate that has been reduced:
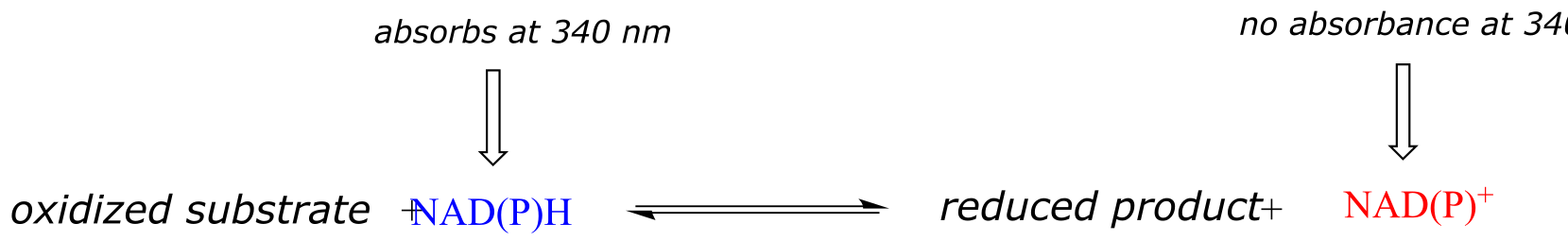
fig 48a
Likewise, a NAD+-dependent dehydrogenase reaction can be followed in real time by monitoring the increase in absorbance at 340 nm as NAD+ is converted to NADH.
Exercise 15.13: You are observing the progress of the (R)-glycerol phosphate dehydrogenase reaction shown in the figure below.
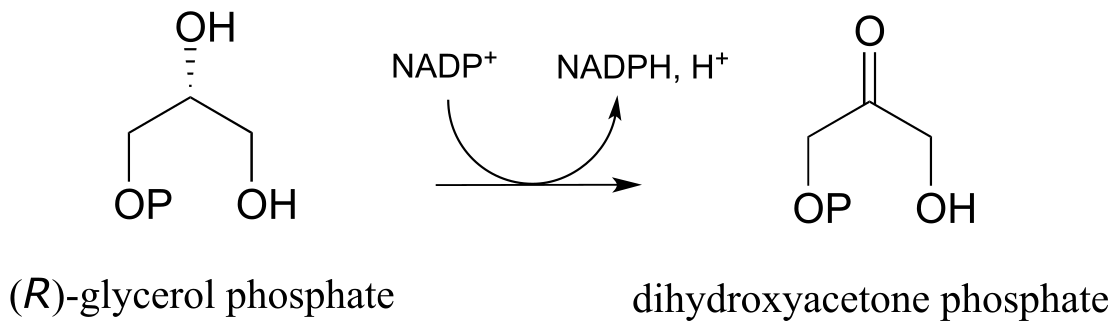
You run the reaction in a quartz cuvette (path length 1 cm) in a total solution volume of 1 mL. You start with 200 μM substrate and 100 μM NADP+ in solution, zero the UV spectrophotometer, then add the enzyme to start the reaction. After 5 minutes, the A340 reading has climbed from 0.000 to 0.096. At this time point:
a) How many moles of substrate have been oxidized?
b) What is the solution concentration of NADP+?
c) The enzyme has a mass of 25 kilodaltons (25,000 g/mol).You added 5 μL of a 2 ng/μL solution of pure enzyme to start the reaction. How many reactions does each enzyme molecule catalyze, on average, per second? (This number is referred to by biochemists as the ‘turnover number’).
Section 15.6: Redox reactions of thiols and disulfides#
A disulfide bond is a sulfur-sulfur bond, usually formed from two free thiol groups.
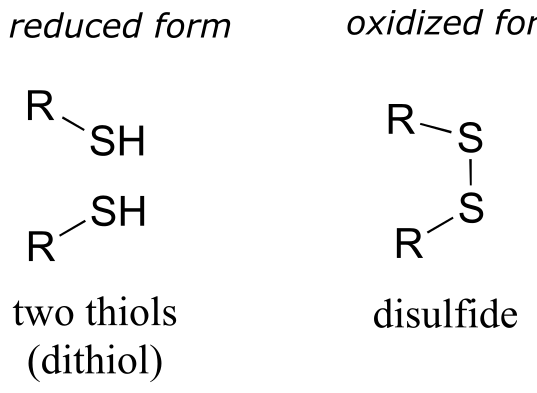
fig 49
The interconversion between dithiol and disulfide groups is a redox reaction: the free dithiol form is in the reduced state, and the disulfide form is in the oxidized state. Notice that in the oxidized (disulfide) state, each sulfur atom has lost a bond to hydrogen and gained a bond to sulfur.
As you should recall from your Biology courses, disulfide bonds between cysteine residues are an integral component of the three-dimensional structure of many extracellular proteins and signaling peptides.
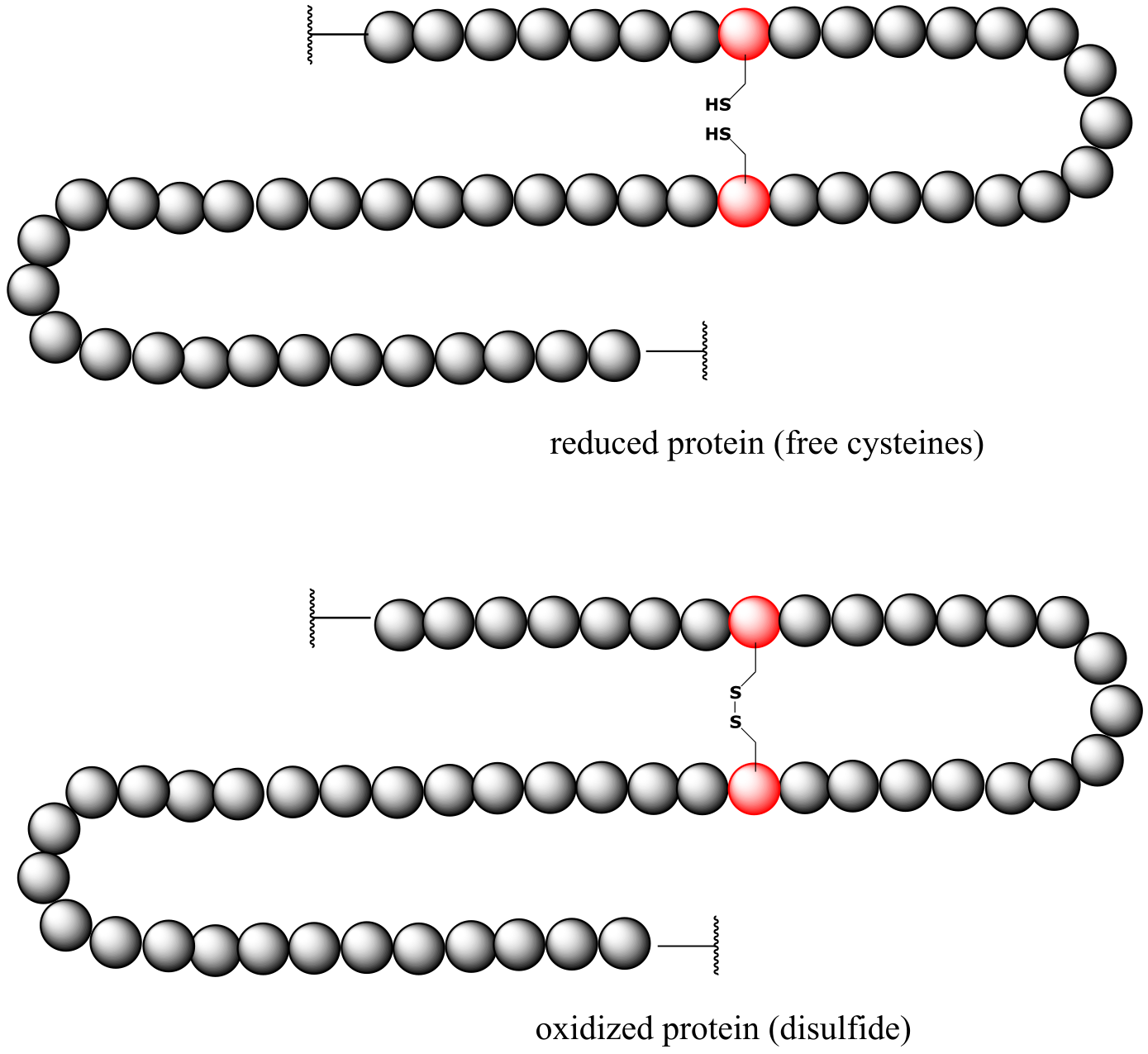
fig 49
fig 49a
A thiol-containing coenzyme called glutathione is integrally involved in many thiol-disulfide redox processes (recall that glutathione was a main player in this chapter’s introductory story about concussion research). In its reduced (thiol) form, glutathione is abbreviated ‘GSH’. In its oxidized form, glutathione exists as a dimer of two molecules linked by a disulfide group, and is abbreviated ‘GSSG’.

fig 50
Disulfide bonds and free thiol groups in both proteins and smaller organic molecules like glutathione can ‘trade places’ through a disulfide exchange reaction. This process is essentially a combination of two direct displacement (SN2-like) events, with sulfur atoms acting as nucleophile, electrophile and leaving group.
Disulfide exchange reaction
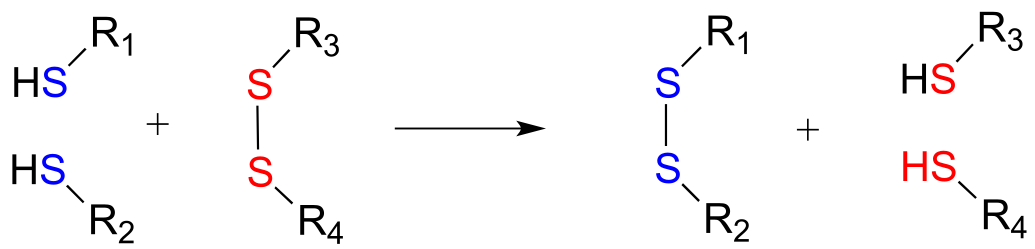
Mechanism:
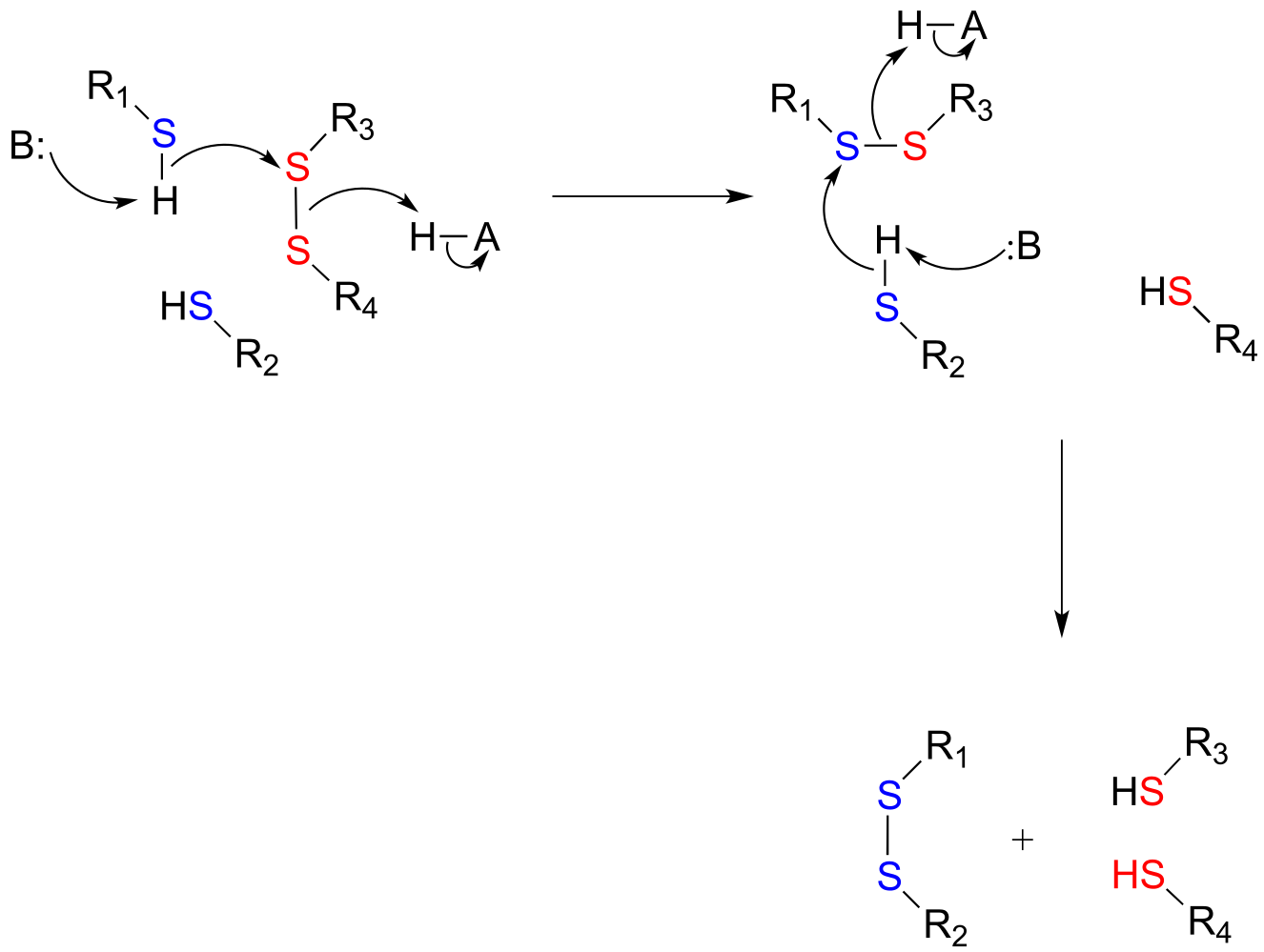
fig 51
In eukaryotes, the cysteine side chains of intracellular (inside the cell) proteins are almost always in the free thiol (reduced) state due to the high concentration of reduced glutathione (GSH) in the intracellular environment. A disulfide bond in an intracellular protein will be rapidly reduced in a disulfide exchange reaction with excess glutathione.
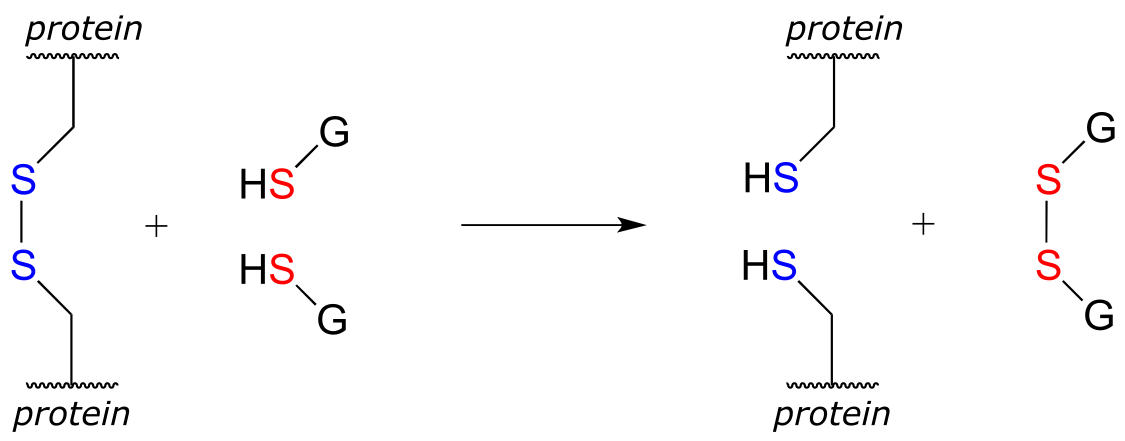
fig 51
The interconversion of free thiols and disulfides is also mediated by flavin in some enzymes.
Flavin-mediated reduction of a protein disulfide bond
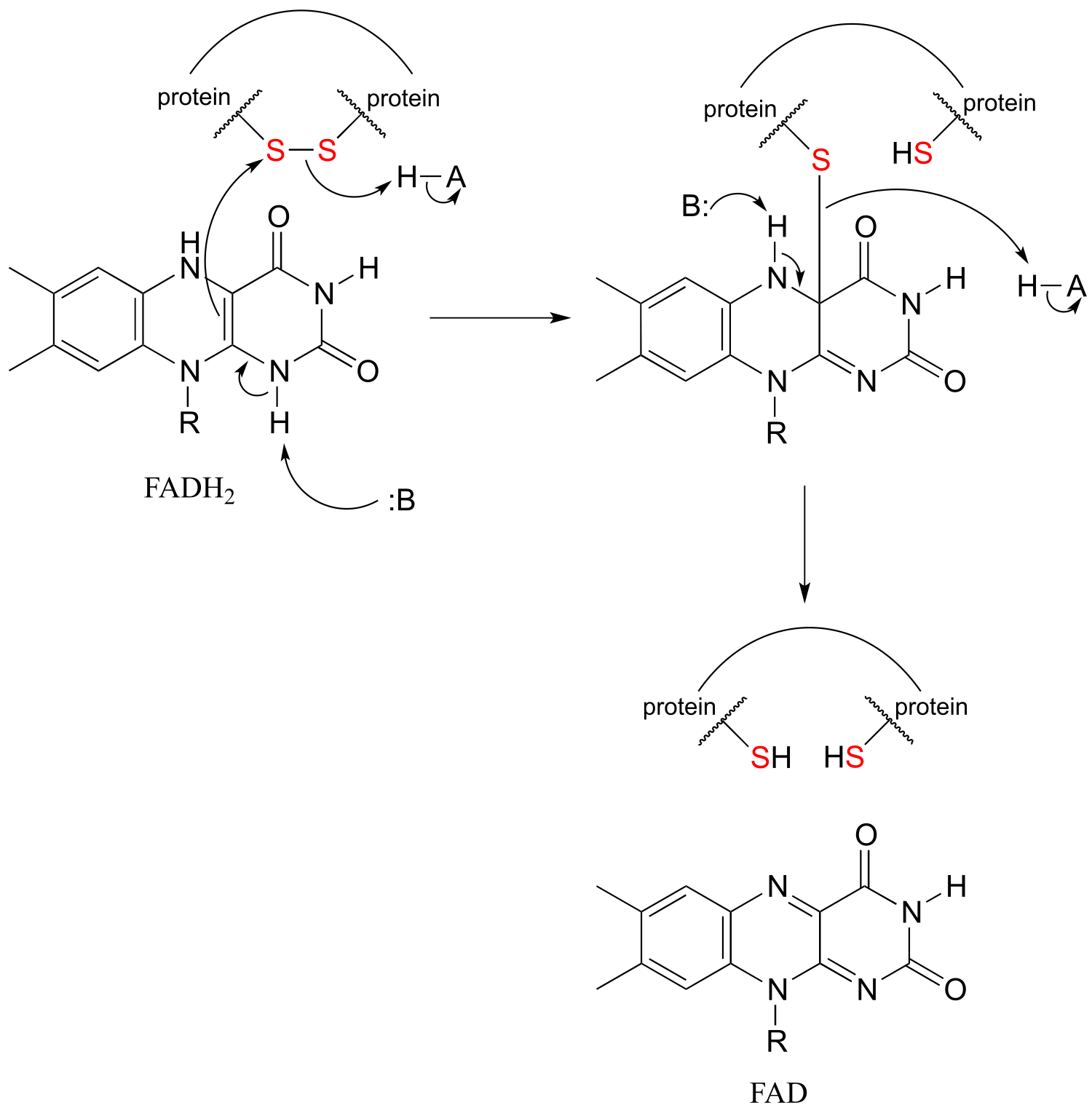
fig51c
Flavin-mediated oxidation of a protein disulfide bond
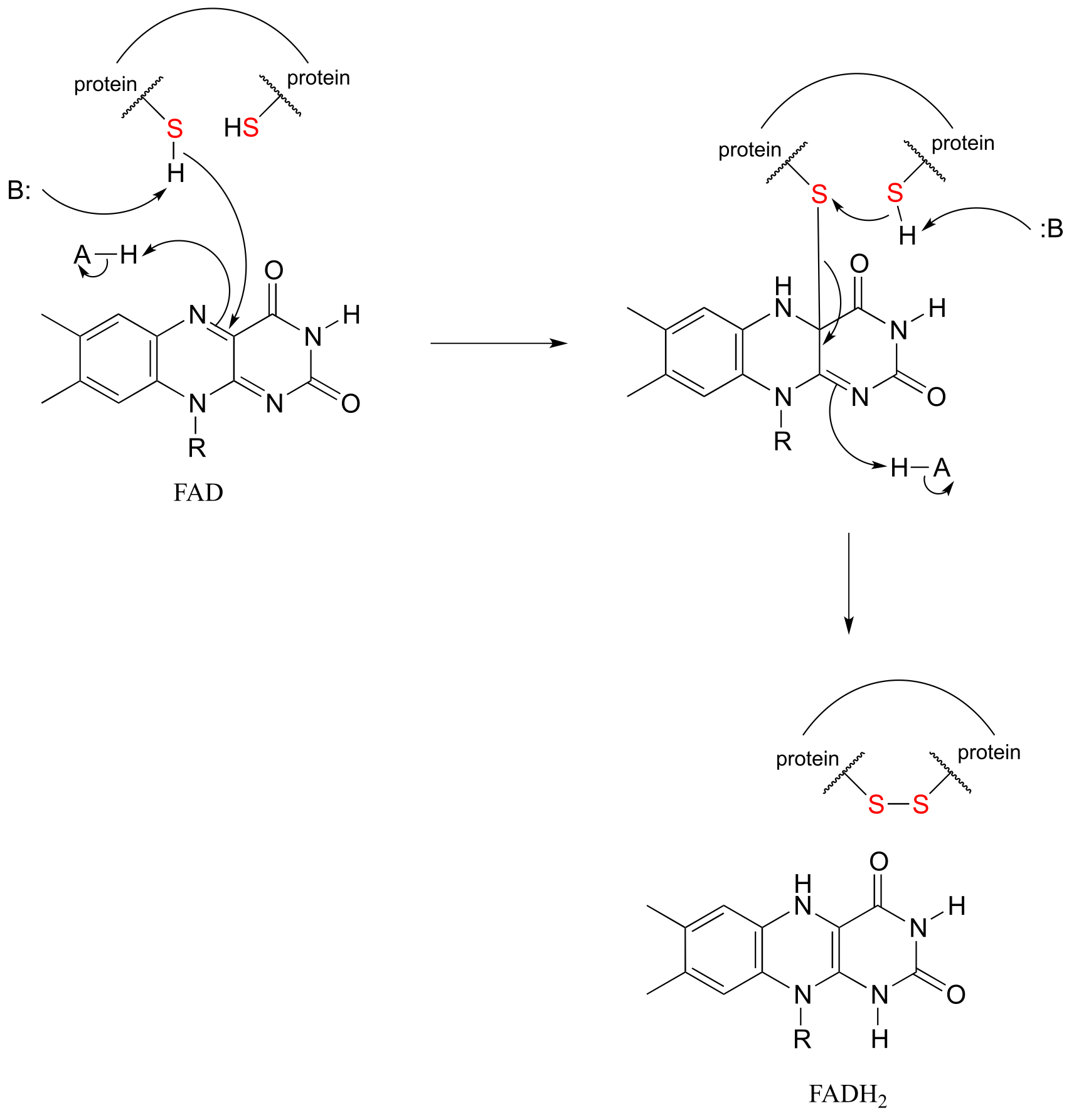
fig 51h
As was stated earlier, a high intracellular concentration of reduced glutathione (GSH) serves to maintain proteins in the free thiol (reduced) state. An enzyme called glutathione reductase catalyzes the reduction of GSSG in a flavin-mediated process, with NADH acting as the ultimate hydride donor.
Gluthione reductase reaction:
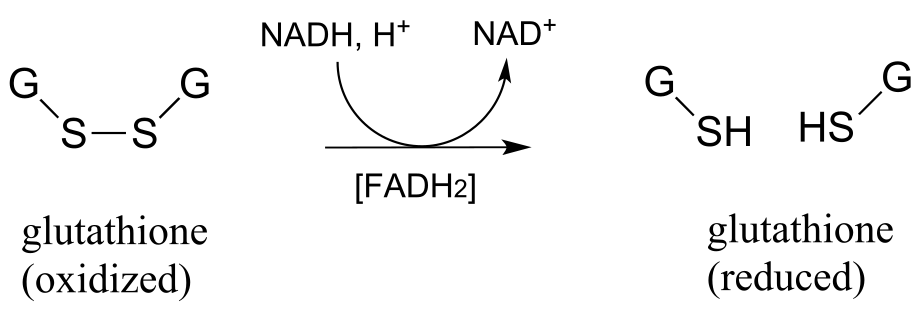
fig 51a
The mechanism for this and other similar reactions is not yet completely understood, but evidence points to an initial thiol-disulfide exchange reaction with a pair of cysteines from the enzyme, (phase 1 below) followed by flavin-dependent reduction of the cysteine-cysteine disulfide (phase 2). Finally, (phase 3) FAD is reduced back to FADH2 by NADH. Frey and Hegeman, Enzymatic Reaction Mechanisms, p. 699
Phase 1: thiol-disulfide exchange (see earlier figure for mechanism):
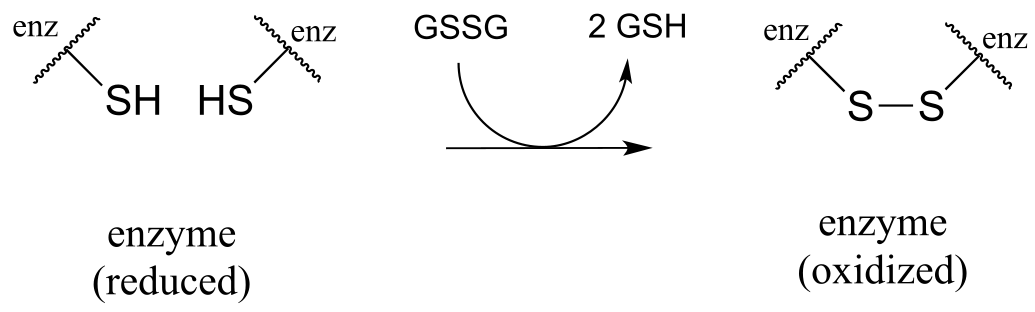
fig 51b
Phase 2: Reduction of protein disulfide by FADH2 (see earlier figure for mechanism)
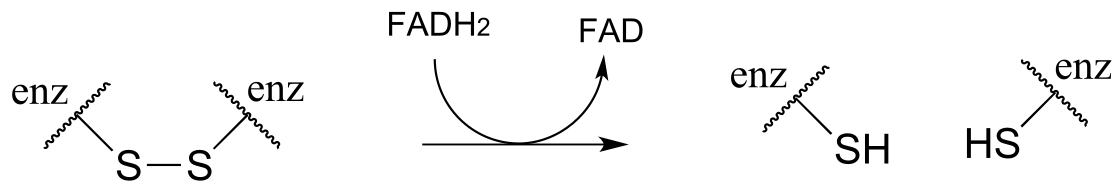
fig 51c
Phase 3: regeneration of FADH2 by NADH (see section 15.4B for mechanism)
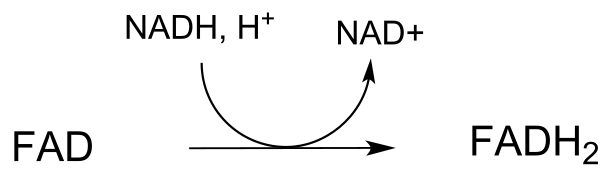
fig 51d
In the biochemistry lab, proteins are often maintained in their reduced (free thiol) state by incubation in buffer containing an excess concentration of β-mercaptoethanol (BME) or dithiothreitol (DTT). These reducing agents function in a manner similar to that of GSH, except that DTT, because it has two thiol groups, can form an intramolecular disulfide in its oxidized form.
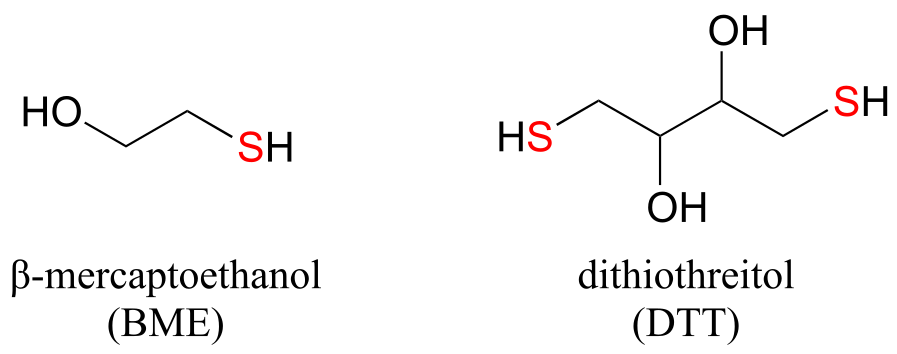
fig 52
Exercise 15.14: Draw structures of the oxidized (disulfide) forms of BME and DTT.
Section 15.7: Flavin-dependent monooxygenase reactions: hydroxylation, epoxidation, and the Baeyer-Villiger oxidation#
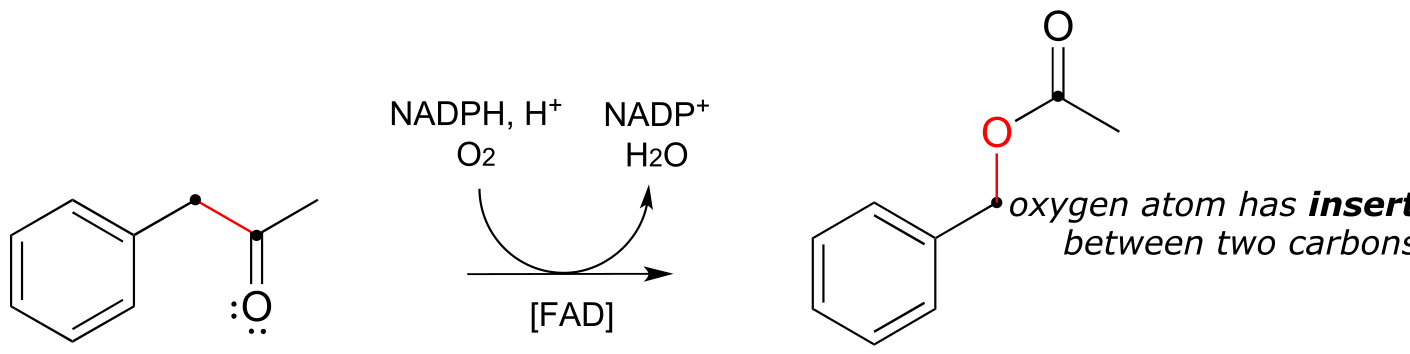
Up to now, the redox reaction examples we have seen have all been either hydrogenation/dehydrogenation transformations or interconversions between free thiols and disulfides. However, there are many important redox reactions in biological chemistry which do not fall under either of these descriptions. Oxygenase enzymes catalyze the insertion of one or two oxygen atoms from molecular oxygen (O2) into an organic substrate molecule. Enzymes which insert a single oxygen atom are called monooxygenases. Below are two examples of biochemical transformations catalyzed by monooxygenase enzymes: one is a hydroxylation, the other is an epoxidation (an epoxide functional group is composed of a three-membered carbon-carbon-oxygen ring - epoxides are somewhat rare in biological organic chemistry but are very common and useful intermediates in laboratory organic synthesis).

fig 56
Dioxygenase enzymes insert both oxygen atoms from O2 into the substrate, and usually involve cleavage of an aromatic ring. Below is an example of a dioxygenase reaction, catalyzed by catechol dioxygenase:
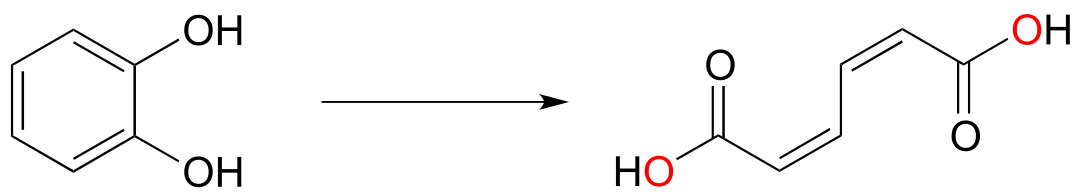
In the reduction direction, reductases remove oxygen atoms, or sometimes other electronegative heteratoms such as nitrogen or halides. For example, DNA deoxyribonucleosides are converted from their corresponding RNA ribonucleosides by the action of reductase enzymes:
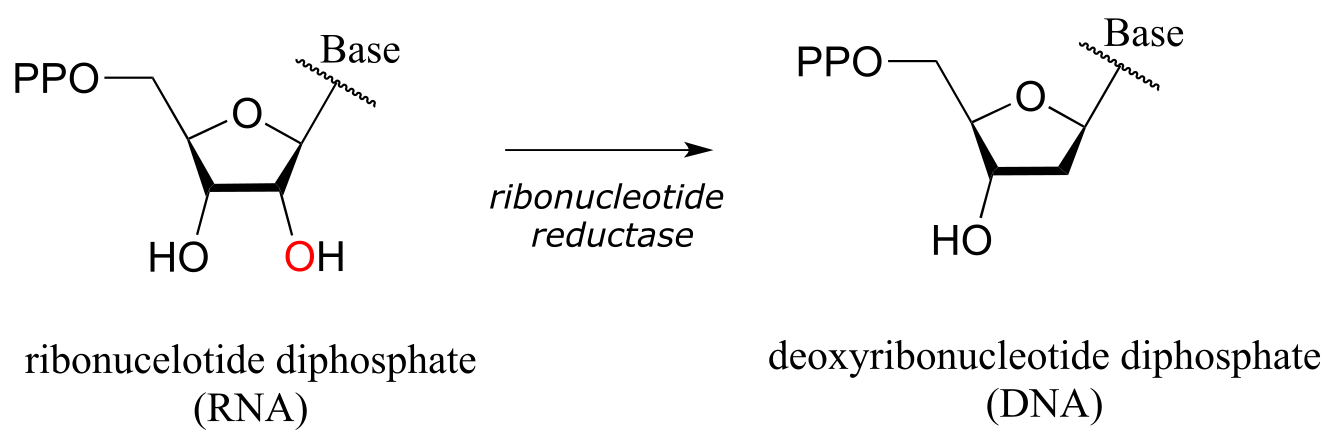
fig 58
Many oxygenase and reductase reactions involve the participation of enzyme-bound transition metals - such as iron or copper - and the mechanistic details of these reactions are outside the scope of our discussion. A variety of biochemical monooxygenase reactions, however, involve flavin as a redox cofactor, and we do have sufficient background knowledge at this point to understand these mechanisms. In flavin-dependent monooxygenase reactions, the key intermediate species is flavin hydroperoxide.
The term ‘peroxide’ refers to a functional group characterized by an oxygen-oxygen single bond. The simplest peroxide is hydrogen peroxide (HOOH) about which we will have more to say below. In flavin hydroperoxide, the peroxide group is linked to one of the carbons of the reactive triple-ring system of the coenzyme. A possible mechanism for the formation of flavin peroxide from FADH2 and molecular oxygen is shown below.
Silverman, R.B. The Organic Chemistry of Enzyme-Catalyzed Reactions, p. 121-122, Scheme 3.33. 2000, Academic Press, San Diego.
Mechanism for the formation of flavin hydroperoxide:
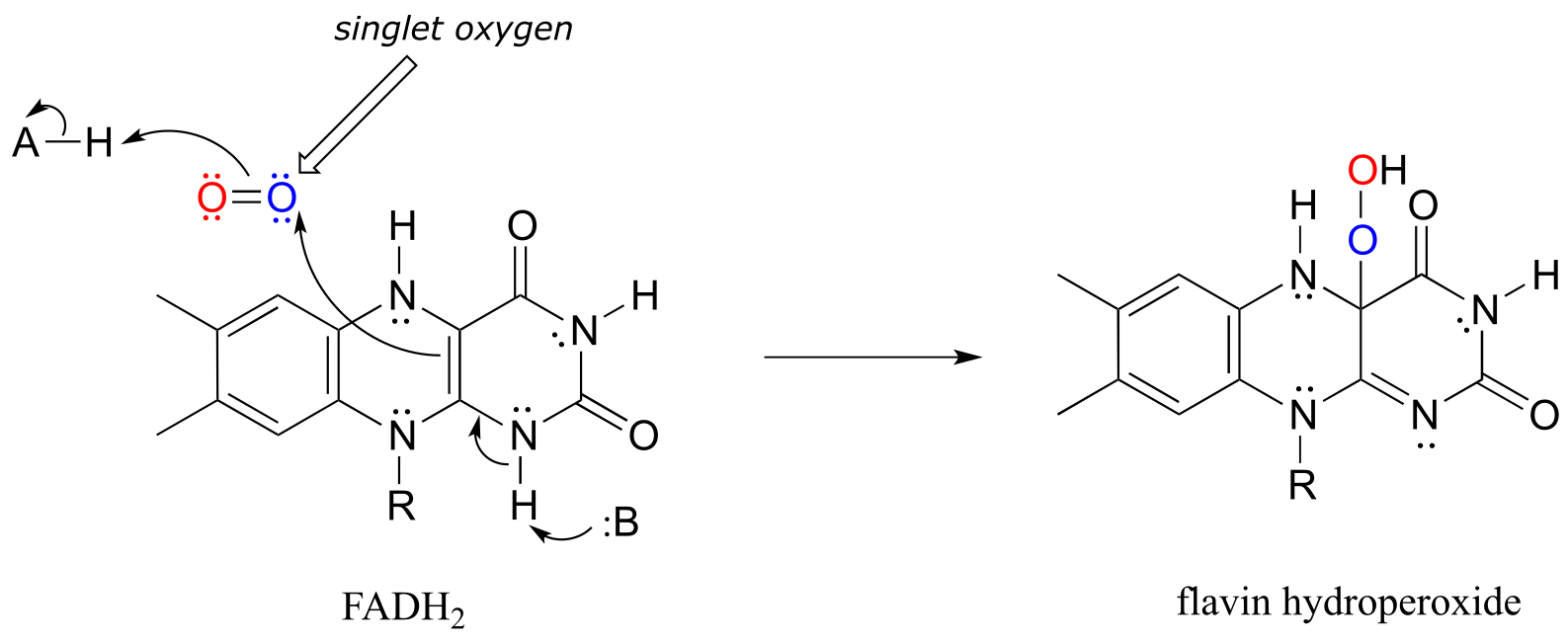
fig 59a
(Note: Implicit in this mechanism is that the molecular oxygen first undergoes spin inversion from the triplet state to the higher energy ‘singlet’ state. You may recall from your general chemistry course that molecular oxygen exists in two states: ‘singlet’ oxygen has a double bond and no unpaired electrons, while ‘triplet’ oxygen has a single O-O bond and two unpaired electrons - a kind of ‘double radical’. Molecular orbital theory - and experimental evidence - show that the triplet state is lower in energy.
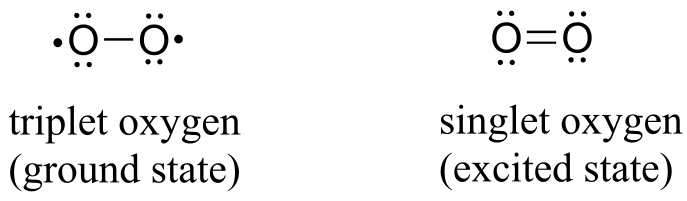
fig 60
The mechanism shown above is one proposed mechanism, another proposal involves triplet oxygen reacting with flavin in a series of radical-intermediate, single-electron steps.)
Flavin hydroperoxide can be thought of as an activated form of molecular oxygen. Peroxides in general are potent oxidizing agents, because the oxygen-oxygen single bond is quite weak: only 138 kJ/mole, compared to 339 kJ/mol for a carbon-carbon bond, and 351 kJ/mol for a carbon-oxygen bond. When the ‘outer’ oxygen of flavin hydroperoxide (red in our figure above) comes into close proximity to the π-bonded electrons of an alkene or aromatic group, the O-O bond will break, leaving an empty orbital on the outer oxygen to be filled by the π electrons - thus, a new carbon-oxygen bond is formed. This is what is happening in step 1 of a reaction in the tryptophan degradation pathway catalyzed by kynurenine 3-monooxygenase. Step 2 completes what is, mechanistically speaking, an electrophilic aromatic substitution reaction (section 14.4) with an peroxide oxygen electrophile.
Mechanism for the flavin hydroperoxide-dependent hydroxylation of kynurenine:
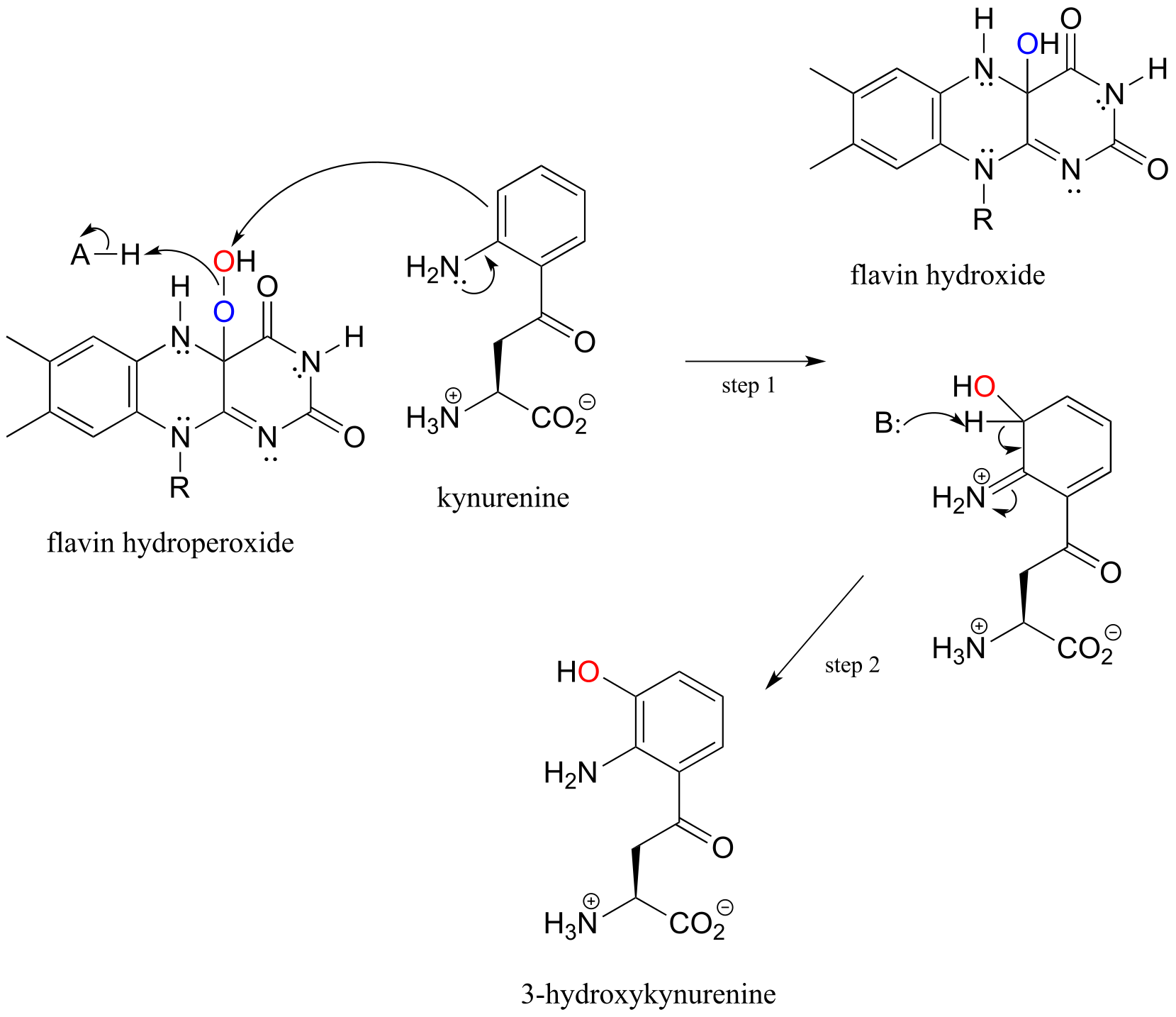
fig 61a

fig 61
Elimination of water from the hydroxyflavin intermediate then leads to formation of FAD (step 3), which is subsequently reduced back to FADH2 by NADH (step 4).
The N-hydroxylation reaction below, which is part of the of the biosynthetic pathway of an iron-binding molecule in the pathogenic bacterium Pseudomonas aeruginosa, is mechanistically similar to the C-hydroxylation reaction we just saw, except that the nucleophile is an amine nitrogen. Note that FADH2 is shown in brackets below the reaction arrow, indicating that reduced flavin participates in the reaction but is not used up - rather it is regenerated in the active site at the end of the reaction cycle.
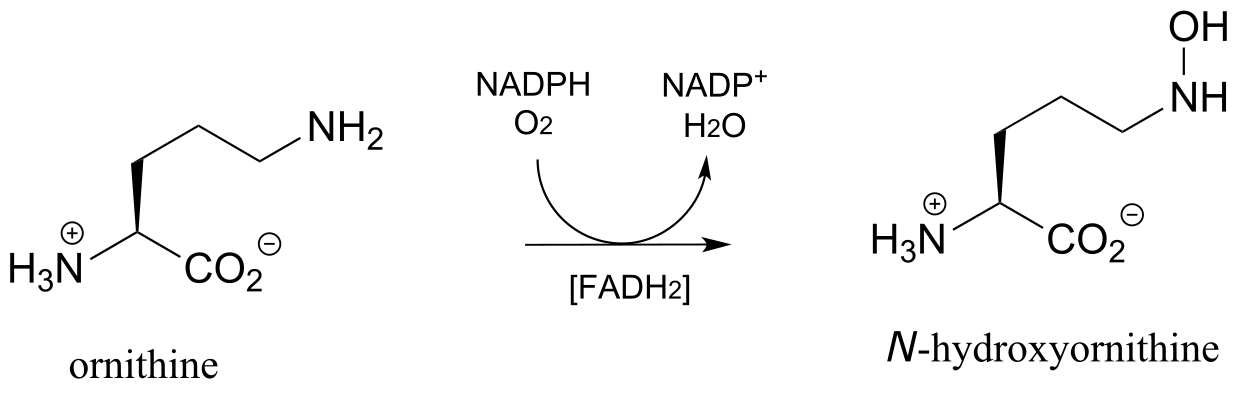
fig 61b
Exercise 15.15: Draw arrows for the N-O bond-forming step in the ornithine hydroxylation reaction above.
Epoxides, characterized by a three-membered ring composed of two carbons and one oxygen, are a very common and useful functional group employed in synthetic organic chemistry. Although rare, there are some interesting epoxide-forming reactions in biochemical pathways, catalyzed by flavin-dependent monooxygenase enzymes.
In a key step in the biosynthesis of cholesterol and other steroid compounds, an alkene is converted to an epoxide in a precursor molecule called squalene. Flavin hydroperoxide also serves as the direct oxidizing agent in this step:
Mechanism for the flavin-hydroperoxide-dependent epoxidation of squalene:
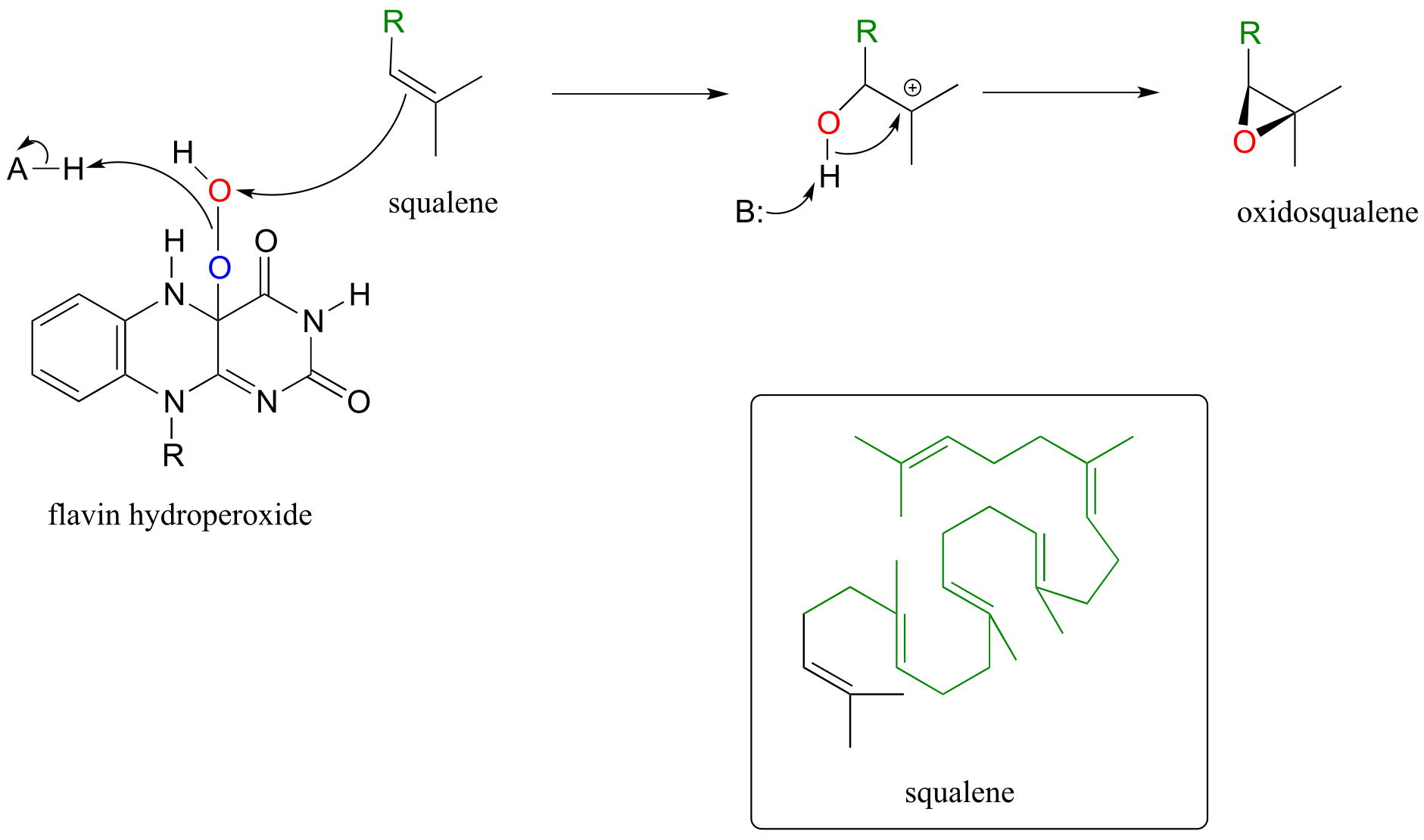
fig 62
Oxidosqualene goes on to cyclize to lanosterol in a complex and fascinating electrophilic reaction which we discussed in section 14.5.
Epoxidation reactions have a parallel in the synthetic organic laboratory, and in fact are very important tools in organic synthesis. In laboratory epoxidations, peroxyacids are the counterpart to flavin hydroperoxide in biochemical epoxidations. meta-chloroperoxybenxoic acid (MCPBA) is a commonly used peroxyacid.
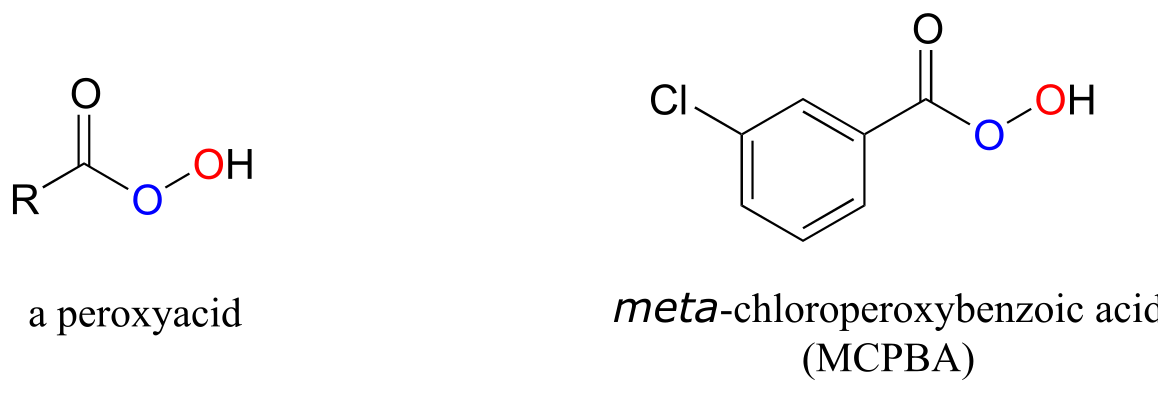
fig 62d
The Baeyer-Villiger oxidation, in which a ketone is converted to an ester through treatment with a peroxide reagent, is an extremely useful laboratory organic synthesis reaction discovered in the late 19th century. Recently, many biochemical examples of Baeyer-Villiger oxidations have been discovered: the reaction below, for example, is catalyzed by a monooxygenase in a thermophilic bacterium:
(Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 13157)
A Baeyer-Villiger oxidation:
Mechanism:
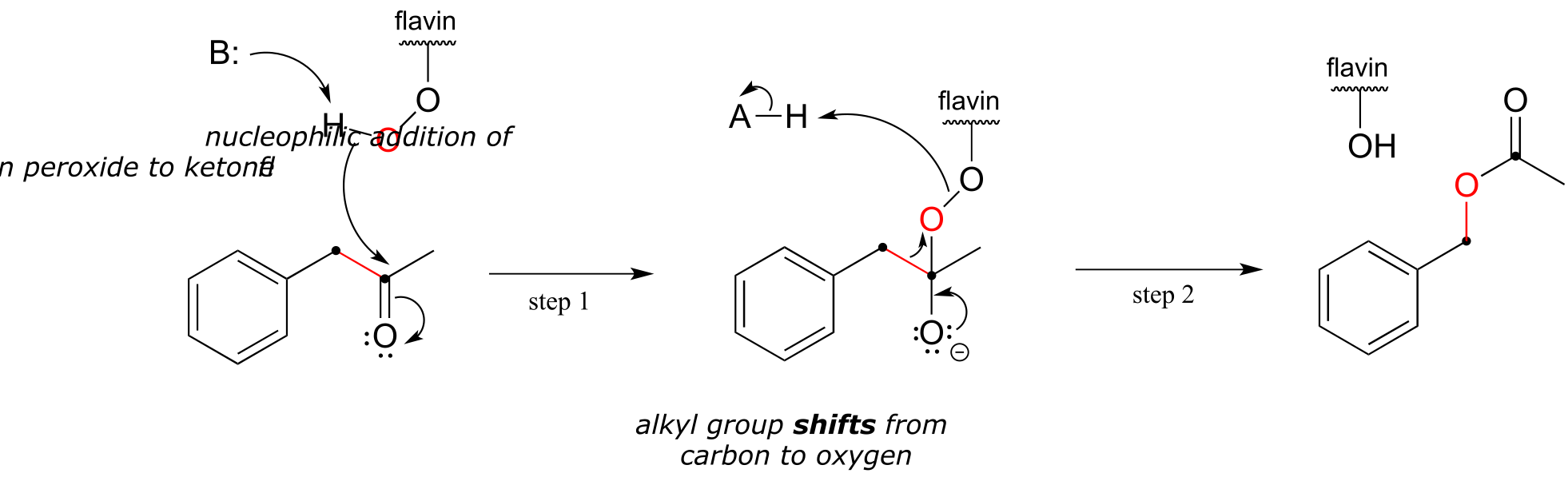
fig 62a
The Baeyer-Villiger mechanism is differs significantly from the hydroxylation reactions we saw earlier, although flavin hydroperoxide (abbreviated in the above figure) still plays a key role. Here, the peroxide oxygen is a nucleophile, rather than an electrophile, attacking the ketone carbonyl in step 1. Step 2 is a rearrangement, similar in many ways to the hydride and alkyl shifts we learned about in section 14.5. The electrons in the red bond in the figure shift over one atom: from the carbonyl carbon to the outer peroxide oxygen. The end result is that an oxygen atom, from O2 via flavin hydroperoxide, has been inserted between the carbonyl carbon and a neighboring methylene (CH2) carbon, forming an ester.
Note that in the reaction mechanism above, the ketone substrate is asymmetric: on one side of the carbonyl there is a benzyl group (CH2-phenyl), and on the other side a methyl group. Note also that it is the benzyl group, not the methyl, that shifts in step 2 of the mechanism. For reasons that are not yet well understood, in Baeyer-Villiger reactions the alkyl group with higher carbocation stability has a higher migratory aptitude: in other words, it has a lower energy barrier for the shifting step.
Exercise 15.16: Draw the product of a hypothetical Baeyer-Villiger reaction involving the same substrate as the above figure, in which the methyl rather than the benzyl group shifts.
Exercise 15.17: Draw the likely major product of a hypothetical Baeyer Villiger reaction starting with 2-methylcyclopentanone as the substrate. Take into account the idea of migratory aptitude.
Below is another example of a Baeyer-Villiger reaction in which a cyclic ketone is oxidized to a lactone (cyclic ester). Notice that oxygen insertion expands the ring from 6 to 7 atoms. This is the third-to-last step in the biosynthesis of the anti-cancer agent mithromycin in some bacterial species (ACS Chem. Biol. 2013, 8, 2466).
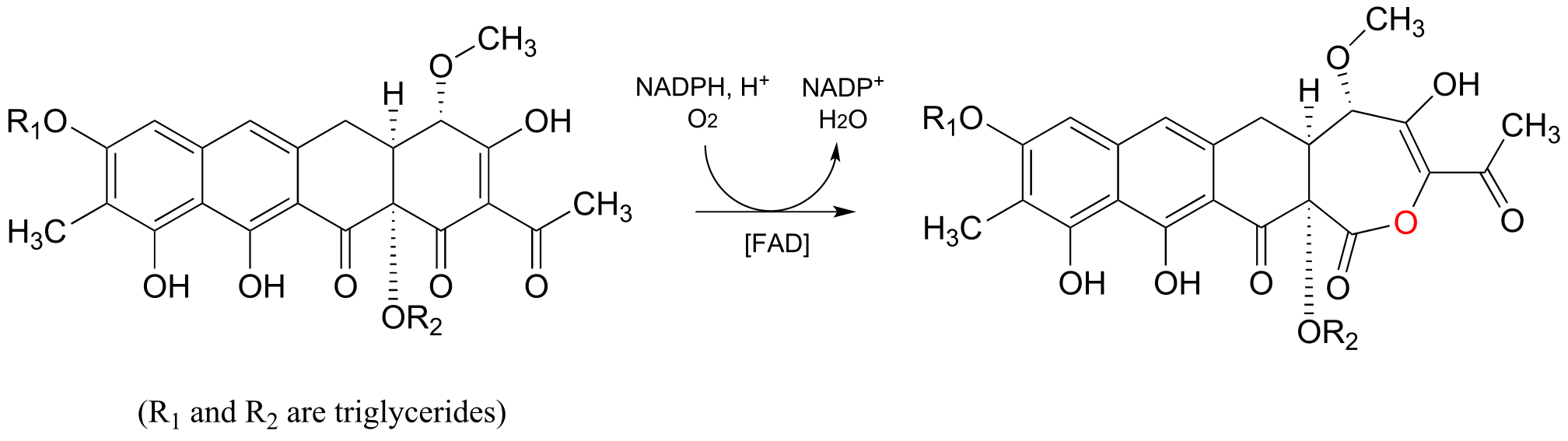
fig 62c
Yet another variety of flavin-dependent monooxygenase, which bears some mechanistic similarity to the Baeyer-Villiger oxidation, is the decarboxylative reaction below from biosynthesis of the plant hormone auxin: (J. Biol. Chem. 2013, 288, 1448)
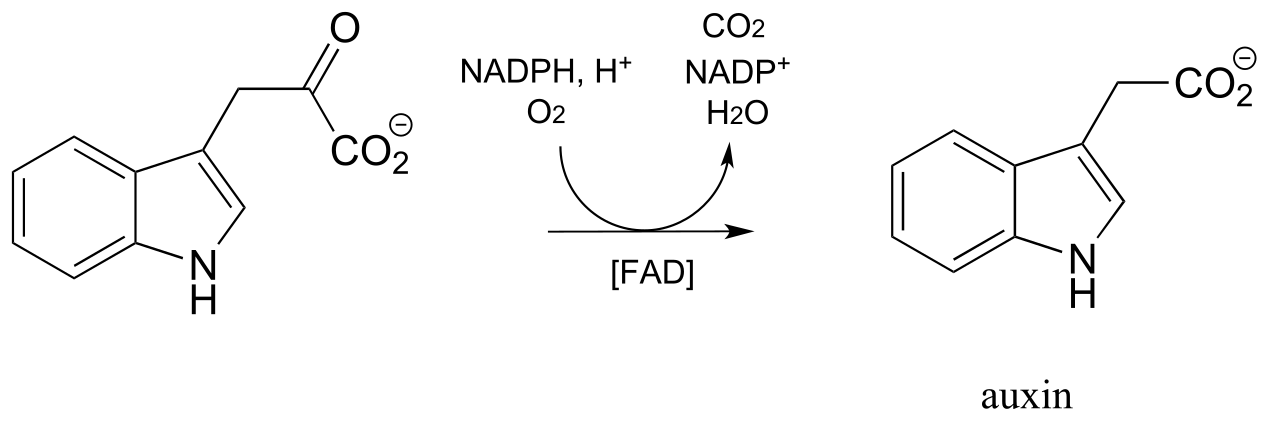
fig 62e
Exercise 15.18: Propose a mechanism for the above reaction, starting with flavin hydroperoxide.
Section 15.8: Hydrogen peroxide as a dangerous Reactive Oxygen Species#
We get our energy from the oxidation of organic molecules such as fat and carbohydrates, as electrons from these reduced compounds are transferred to molecular oxygen, thereby reducing it to water. Reducing O2, however, turns out to be a hazardous activity: harmful side products called reactive oxygen species (ROS) are inevitably formed in the process. Recall from the story introducing this chapter that ROS appear to play an important role in the damage that occurs to the brain immediately after a concussion.
Hydrogen peroxide, HOOH, is an ROS. Recall peroxides are potent oxidizing agents due to the weakness of the O-O single bond. It is this same weak bond that causes hydrogen peroxide to be dangerous when produced in our bodies, as it can react spontaneously with oxygen or nitrogen nucleophiles and π bonds.
Peroxide formed as a by-product of our metabolism is particularly harmful when it oxidizes DNA bases. In just one of many known examples of oxidative damage, the DNA base cytosine is oxidized to thymine glycol in the presence of hydrogen peroxide. Although mechanistic details for reactions such as these are not yet well understood, one possibility is electrophilic addition:
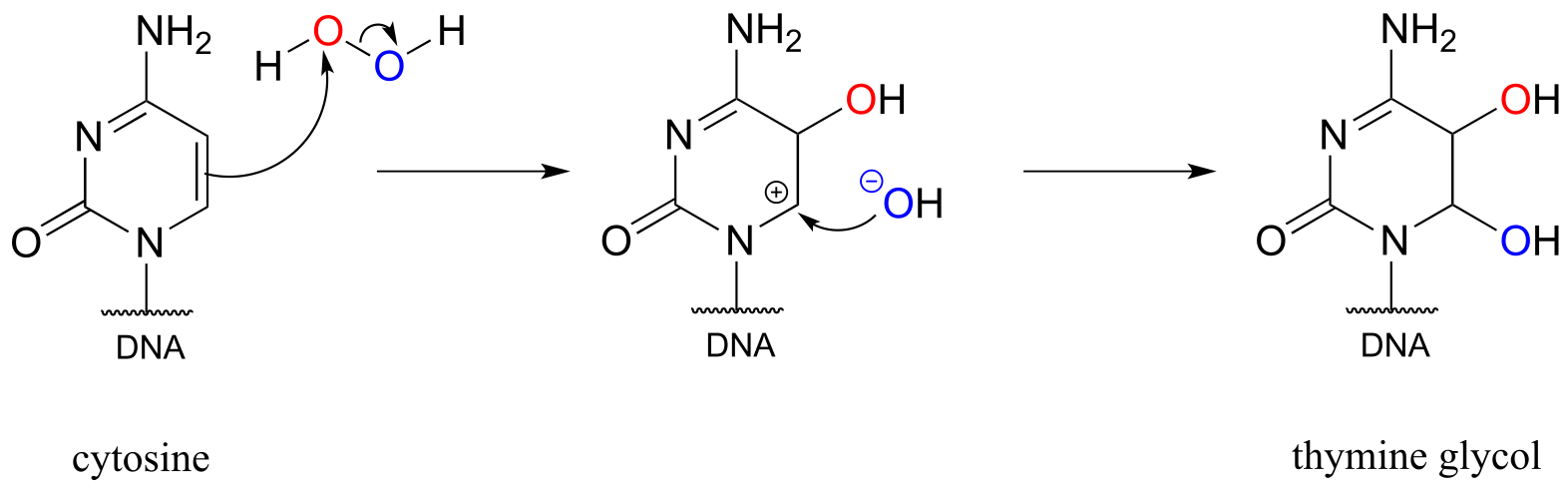
fig 53
Our bodies have evolved ways to dispose of the harmful reactive oxygen species that are continuously being formed (the only way to stop the production of ROS is to stop breathing oxygen!). Glutathione peroxidase is a remarkable enzyme in that its active site contains selanocysteine, a modified cysteine residue in which the side chain sulfur is replaced by selenium (selenium is very toxic, but we do need a very small amount of it in our diet). Look at a periodic table: selenium is below oxygen and sulfur in the same column. If you think back to the vertical periodic trends in nucleophilicity (section 8.2B), you’ll recall that just as a thiol is a better nucleophile than an alcohol, a selanol (RSeH) is even more nucleophilic than a thiol. Moreover, the vertical periodic trend in acidity (section 7.3A) tells us that a selenol should be more acidic than a thiol - in fact, the pKa of a selenocysteine is about 5.5, meaning that it is mostly in its deprotonated state at physiological pH, making it even more nucleophilic.
Glutathione peroxidase very efficiently catalyzes the reduction of hydrogen peroxide to water and the oxidation of glutathione (GSH) to GSSG, beginning with nucleophilic attack by the enzymatic selanocysteine on a peroxide oxygen. The intermediates in this process are shown below: each step can be thought of as a concerted nucleophilic displacement similar to those that take place in a disulfide exchange reaction.
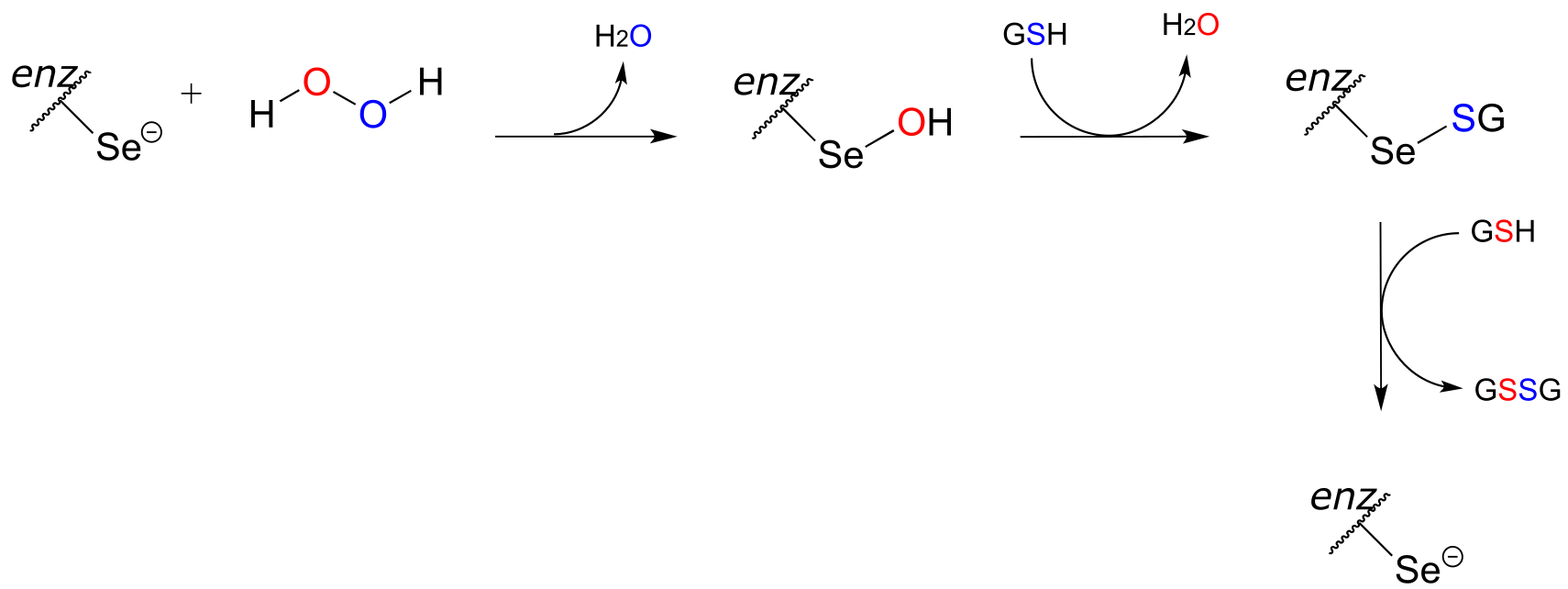
fig 54
Eur. J. Biochem. 1983, 133, 51
Biochem. et Biophys. Acta 2009, 1790, 1486
Key learning objectives for this chapter#
Before moving on to the next chapter, you should be able to:
Recognize when an organic molecule is being oxidized or reduced, and distinguish between redox and non-redox organic reactions.
Draw complete mechanisms for the following reaction types, including the structure of the reactive part of the redox coenzyme (it is strongly recommended that you commit to memory the structures of the reactive parts of the nicotinamide and flavin coenzymes).
oxidation of an alcohol to an aldehyde or ketone
oxidation of an amine to an imine
oxidation of an aldehyde to a carboxylic acid derivative (usually a thioester or carboxylate)
oxidation of an alkane to an alkene at the α,β position relative to a carbonyl or imine
reduction of an aldehyde or ketone to an alcohol
reduction of an imine to an amine
reduction of a carboxylic acid derivative to an aldehyde
reduction of an α,β - conjugated alkene to an alkane
oxidation of two thiol groups to a disulfide in a disulfide-exchange type reaction
reduction of a disulfide group by flavin
flavin hydroperoxide-dependent hydroxylation, epoxidation, and Baeyer-Villiger reactions
reduction of FAD (or FMN) to FADH2 (or FMNH2) by NAD(P)H.
spontaneous oxidation of an alkene group in a biomolecule by hydrogen peroxide
reduction of hydrogen peroxide by glutathione peroxidase
In addition, you should be able to draw complete mechanisms for hydrogenation-dehydrogenation and disulfide exchange reactions that we have not yet seen specific examples of, based on your understanding of the chemistry involved in these reaction types and organic reaction patterns in general. Several exercises and end-of-chapter problems provide opportunities practice with inferring and drawing mechanisms of less familiar redox reactions.
Given a multistep pathway diagram, you should be able to recognize the transformations taking place and fill in missing intermediate compounds or reagents (problem 15.5 is an example of this type).
You should be working on gaining proficiency at solving multi-step pathway elucidation problems, such as those at the end of this chapter’s problem section.
Problems#
P15.1: Show a mechanism for each of the redox reactions below. Do not abbreviate the reactive parts of the redox coenzyme.
a)
b)
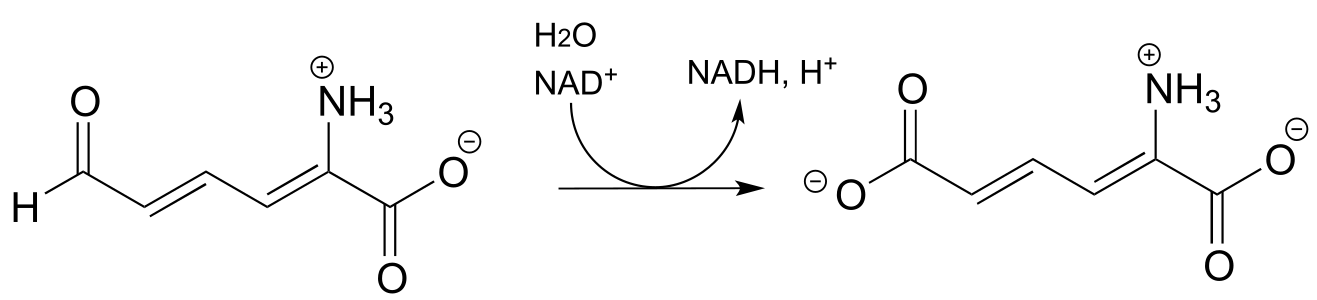
c)
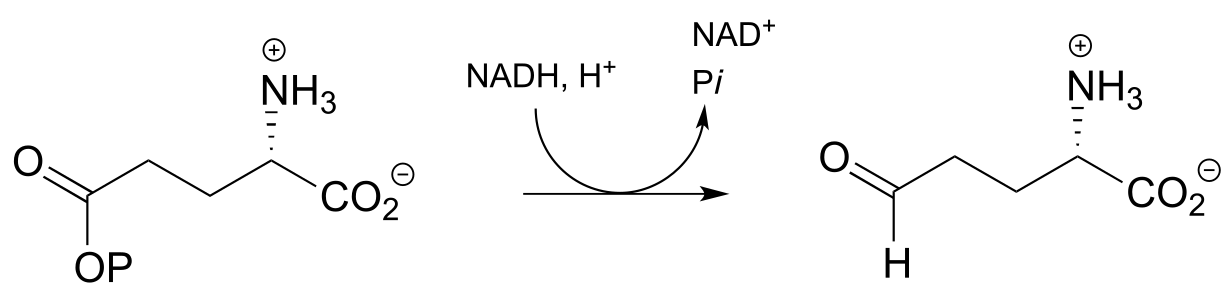
d)
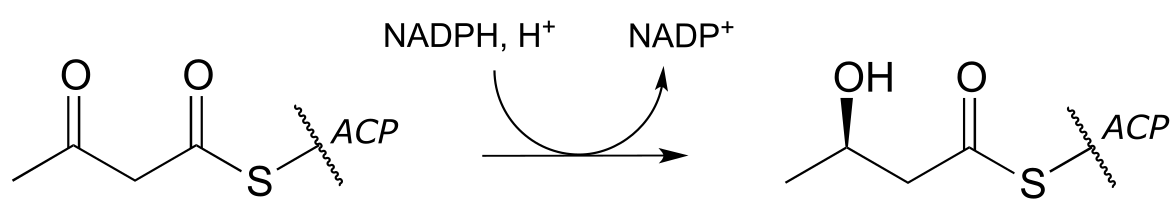
e)
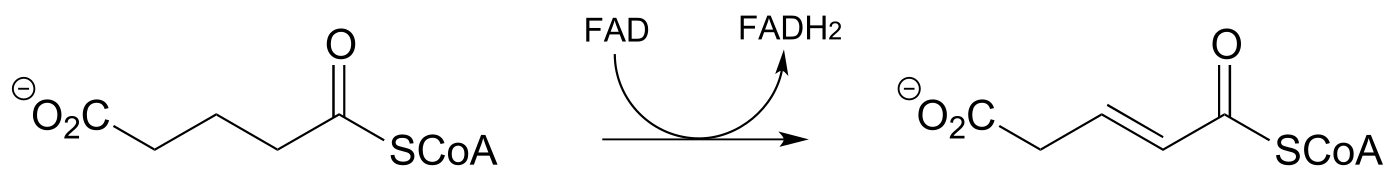
f)
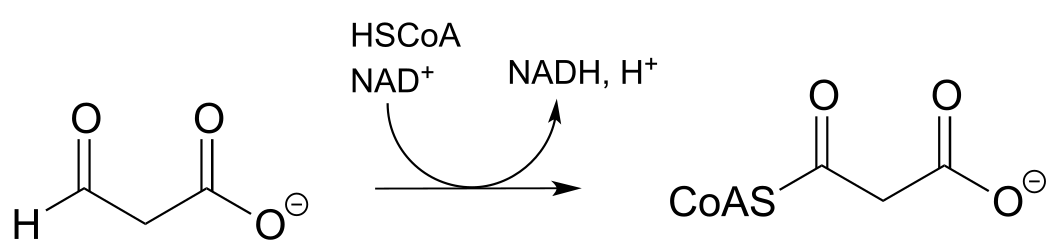
g) (regeneration of reduced flavin by NADH):
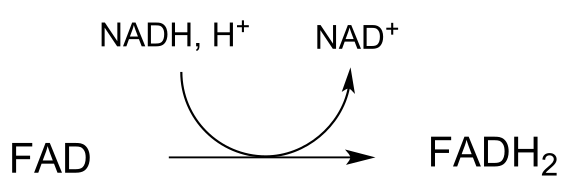
h) In reaction (a), near which face of the substrate is the cofactor bound in the active site?
i) ) In reaction (d), near which face of the product is the cofactor bound in the active site?
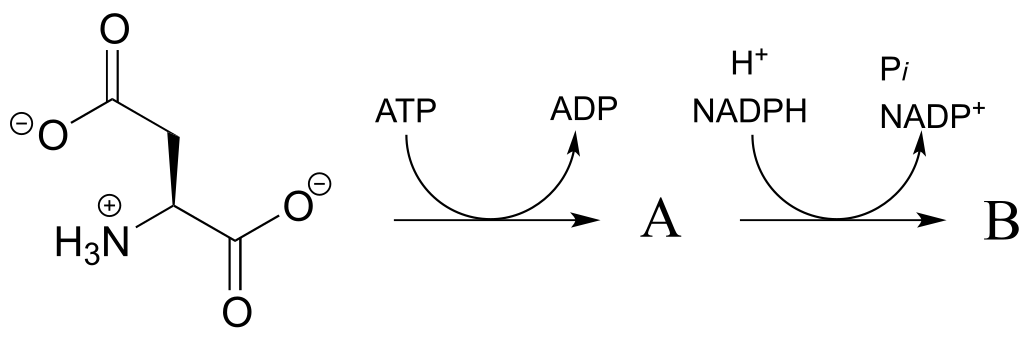
P15.2: In the reactions below ((EC 2.7.2.4; EC 1.2.1.11) , the side chain of aspartate is altered, but the main peptide chain is not affected. Show the most probable structure of species A and B.
P15.3: Propose complete mechanisms for the following reactions.
a) (EC 1.3.1.26, from lysine biosynthesis)
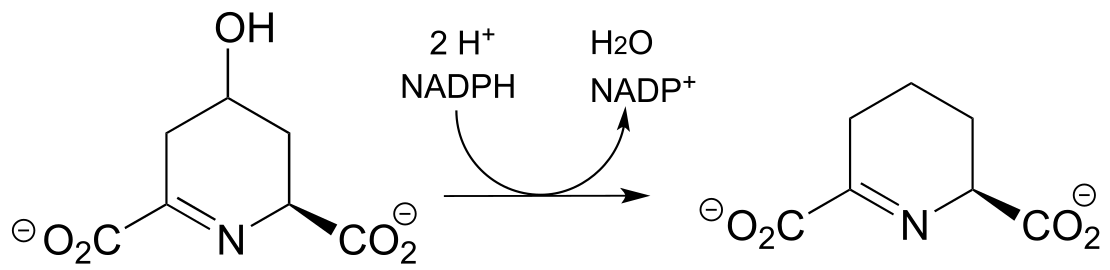
(
b) (EC 1.1.1.205, from guanosine ribonucleotide biosynthesis) The mechanism involves a covalent cysteine-linked enzyme-substrate intermediate.
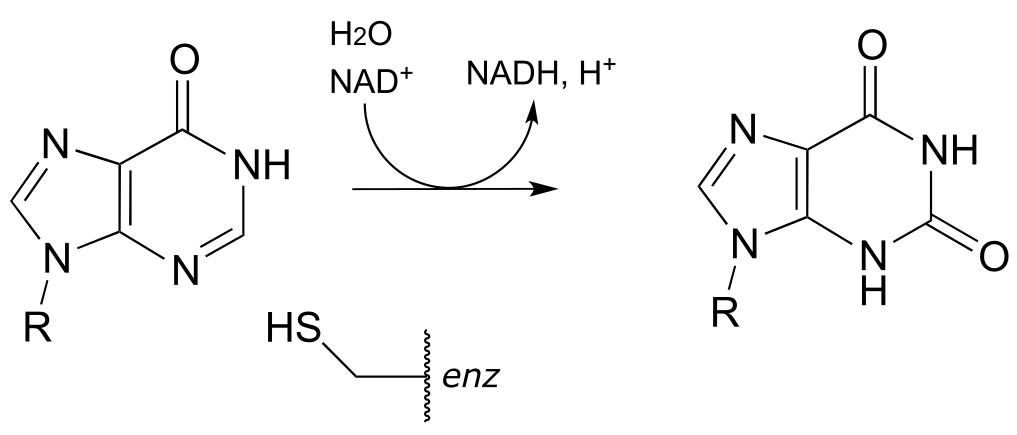
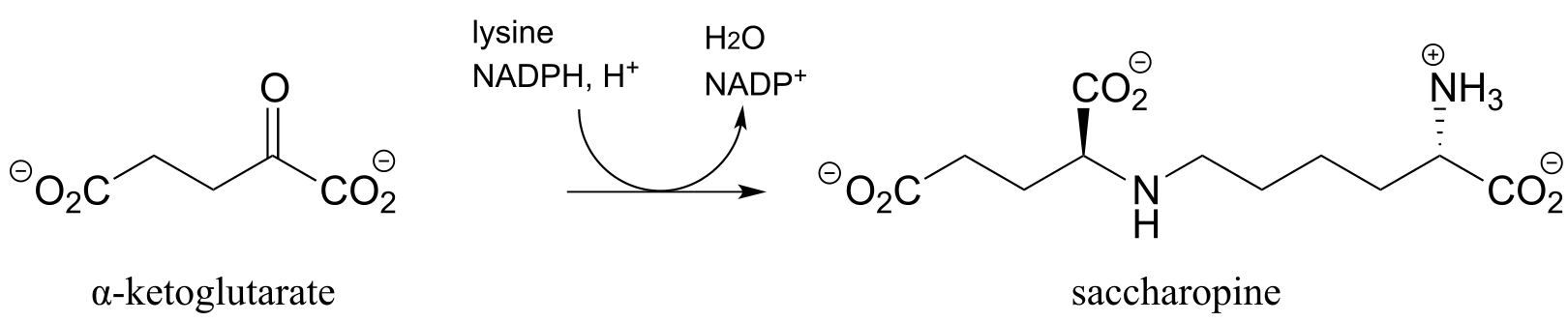
P15.4: The first step in the lysine degradation pathway is a reductive condensation with α-ketoglutarate to form an intermediate called saccharopine. (EC 1.5.1.8)
a) Propose a mechanism for this transformation.
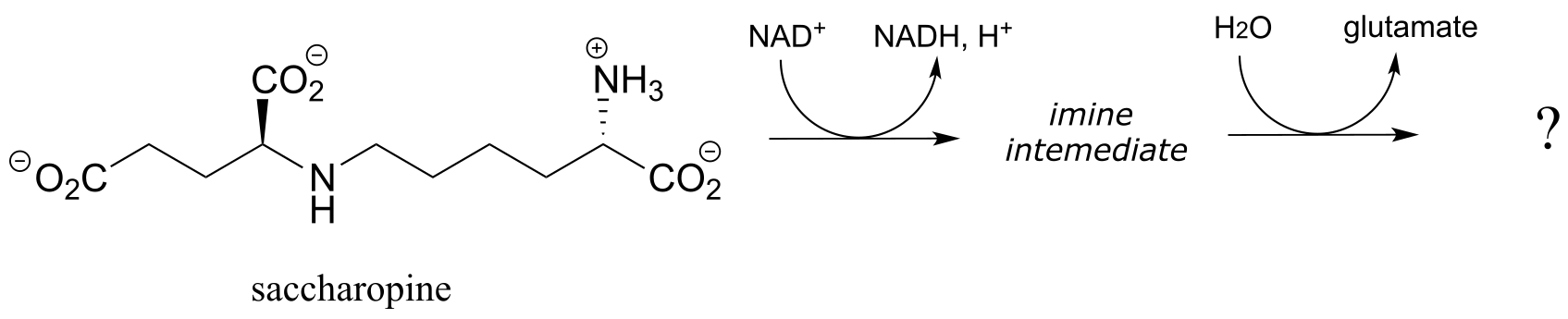
b) Saccharopine (see part (a) above) is then broken up to yield glutamate and a second product that contains an aldehyde group. Predict the structure of this second product, and propose a likely mechanism for the reaction which involves and imine intermediate.
(EC 1.5.1.10
(
**
**
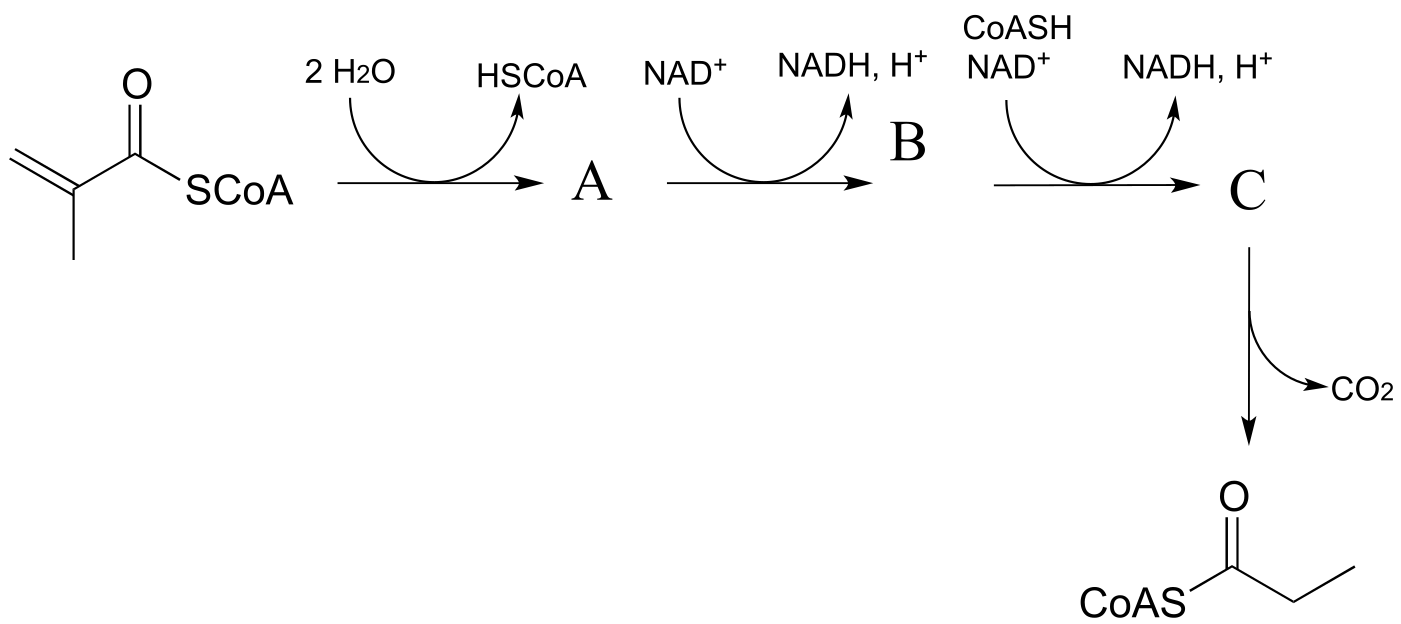
P15.5: Predict the structures of species A and B in the pathway below.
P15.6: Bilirubin, the molecule responsible for the yellowish color of bruises, is formed from the NADPH-dependent hydrogenation of a double bond in biliverdin (EC 1.3.1.24), which is a product of heme degradation (heme is an iron-containing coenzyme in the oxygen-carrying blood protein hemoglobin). Draw a likely mechanism for this reaction.
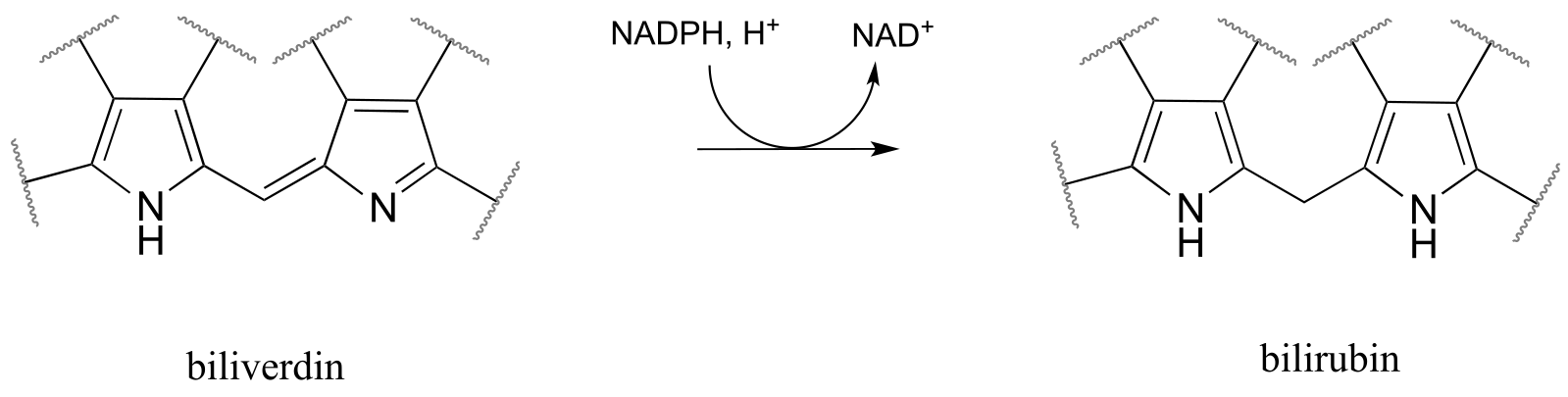
P15.7: Draw a likely mechanism for each of the reactions below.
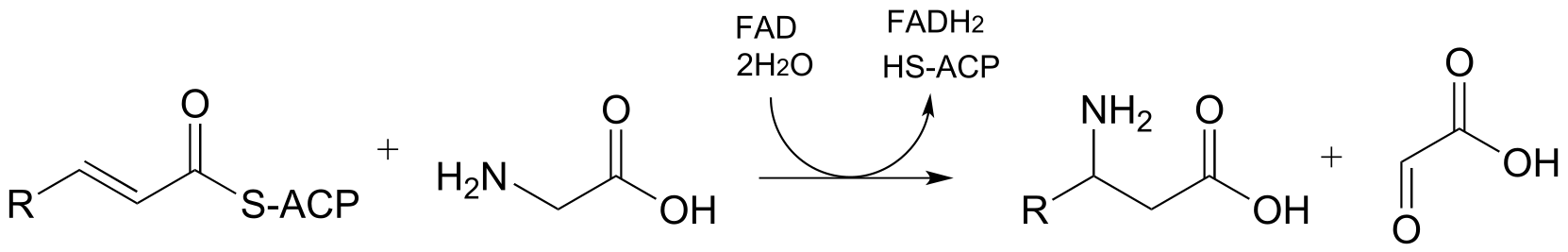
a) From the oxidation of polyunsaturated fatty acids: (J. Biol. Chem 2005, 280, 3068)
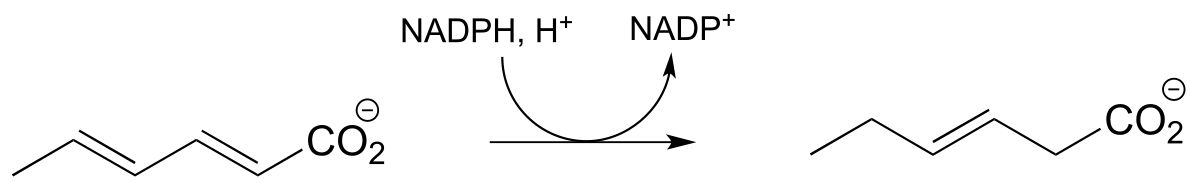
b) (ChemBioChem 2013, 14, 1998, Scheme 2
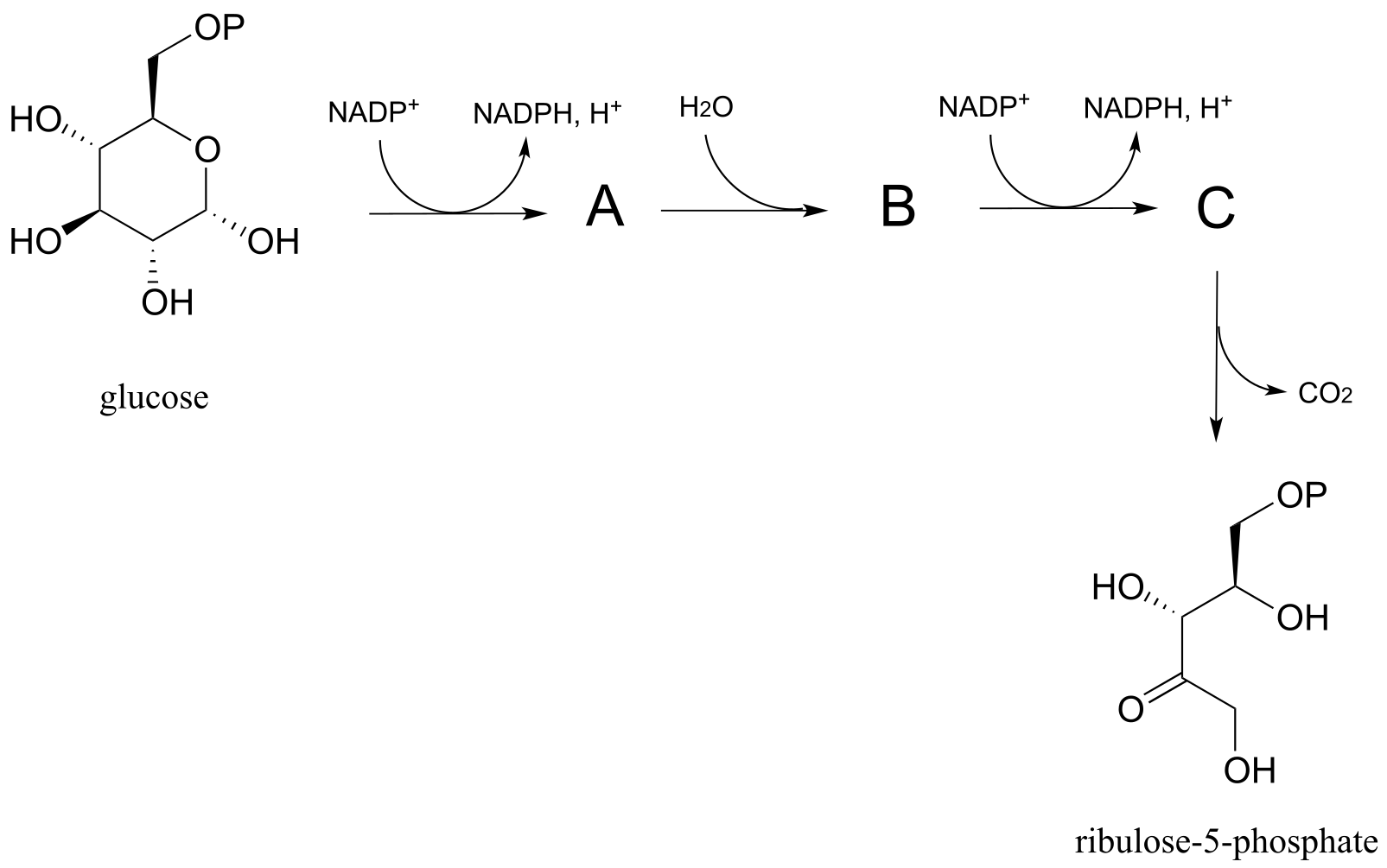
P15.8: Predict the structures of pathway intermediates A, B, and C:
P15.9: An enzyme called DsbA (EC1.8.4.2) is responsible for the formation of disulfide bonds in bacterial proteins. The process - which can be thought of as a ‘disulfide exchange’, involves the cleavage of a disulfide bond between two active site cysteines in DsbA. It is accomplished through two successive SN2 displacements.
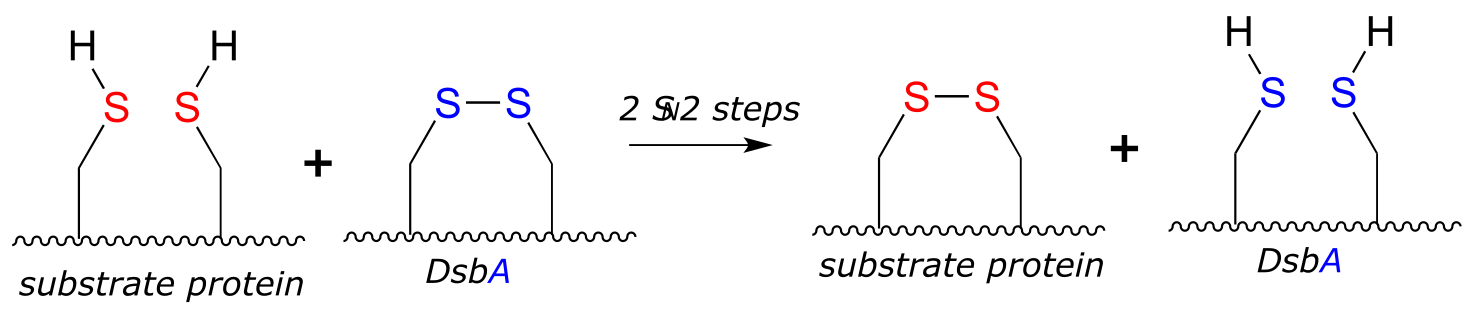
Biochemistry 2000*, 39*, 6732
DsbA is then returned to it’s starting (disulfide) state through a second disulfide exchange reaction with another protein called DsbB:
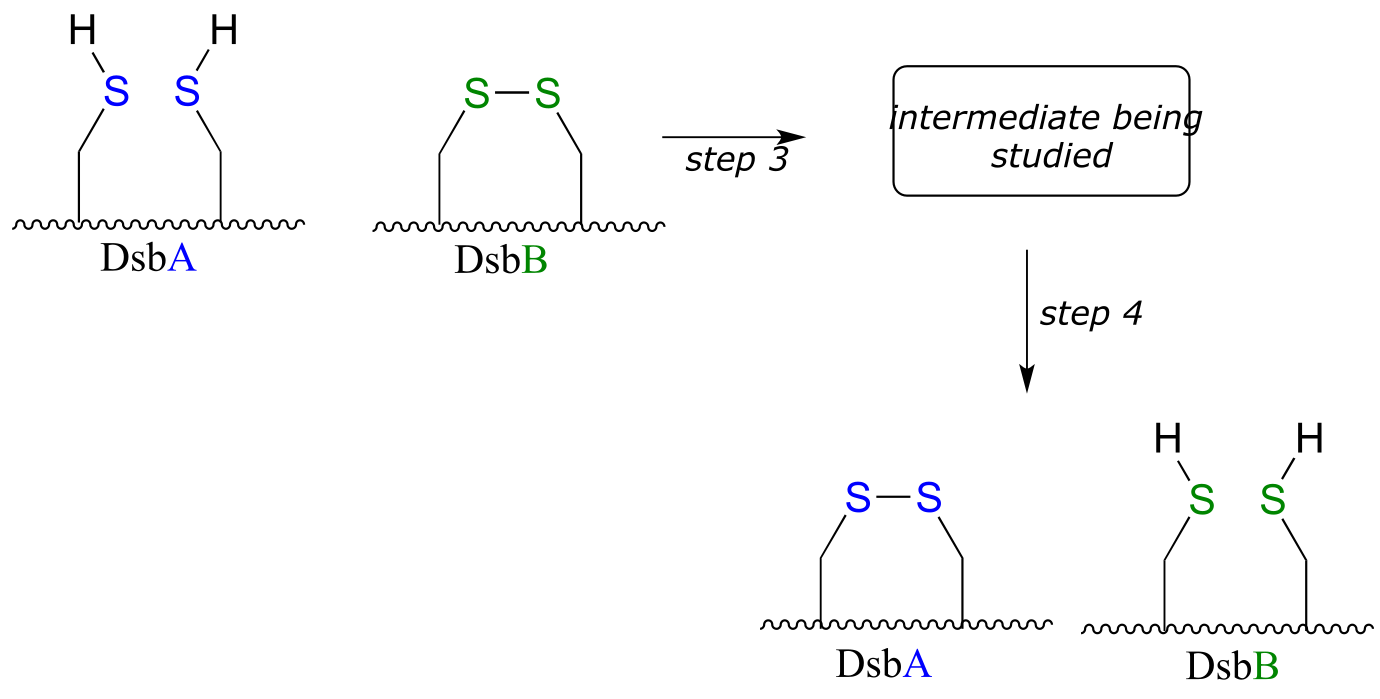
Scientists were interested in studying the intermediate species formed in step 3, but found that it is very short-lived and difficult to isolate. In order to address this problem, they ran the reaction with a synthetic analog of DsbB that contained an unnatural bromoalanine amino acid in place of one of the active site cysteines.
a) Draw a complete mechanism for the disulfide exchange reaction between DsbA and DsbB.
b) Show how the bromoalanine-containing DsbB analog allowed for the isolation of an intermediate that resembles the true, short-lived intermediate.
(J. Am. Chem. Soc. 2004, 126, 15060; J. Nat. Prod. 2001, 64, 597).
P15.10:
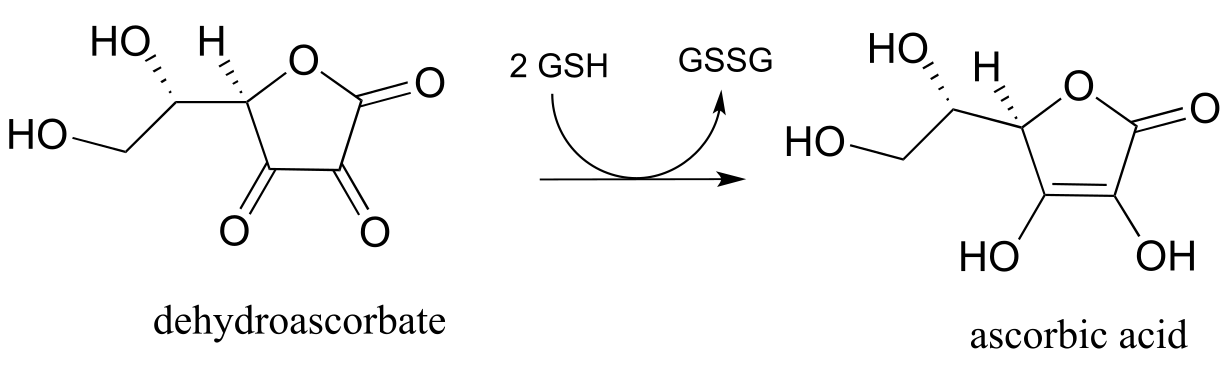
a) In chapter 16 we will learn how ascorbate (vitamin C) acts as a ‘radical scavenger’ antioxidant to protect our cells from damage by free radical species. When ascorbate scavenges a radical, it ends up being converted to dehydroascorbate . One possible metabolic fate of dehydroascorbate is to be recycled back to ascorbate through an enzyme-free reaction with glutathione. Biochemistry 1999, 38, 268.
probfig10a
Suggest a likely mechanism for the enzyme-free reaction.
b) In the introduction to chapter 16, we will learn that most animals - but not humans - are able to synthesize their own ascorbate. Humans cannot synthesize vitamin C because we lack the enzyme for the final step in the biosynthetic pathway, gulonolactone oxidase:
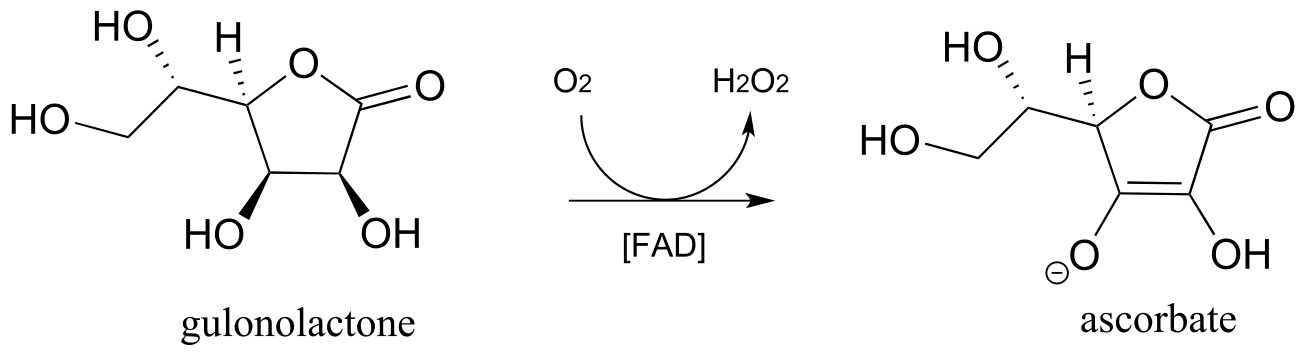
This enzyme uses FAD as an oxidizing agent, and FADH2 is oxidized back to FAD at the end of the catalytic cycle by molecular oxygen, with hydrogen peroxide as a side product. Draw out a likely mechanism showing how gulonolactone is converted to ascorbate, and how FAD is regenerated.
c) Artemisinin is a naturally occurring compound with demonstrated antimalarial properties. It is thought to act by depleting the malaria-causing microbe’s store of reduced flavin, thus disrupting the redox balance. The relevant reaction is shown below:
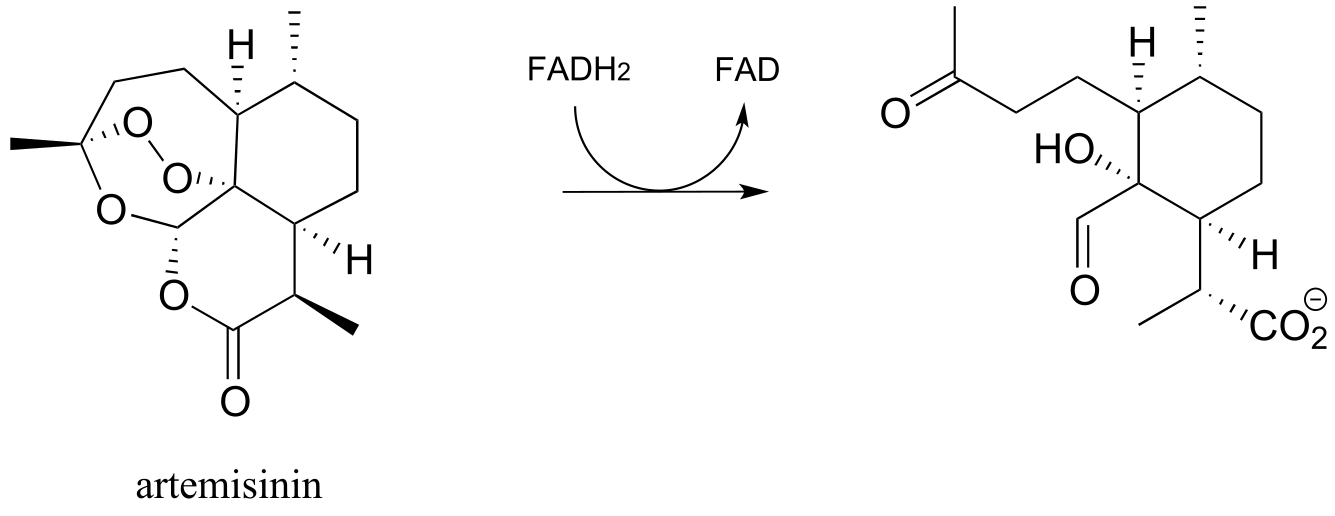
Draw mechanistic arrows for the step as shown above. (Molecules 2010, 15, 1705)
d) Draw the mechanistic step in the reaction below in which the C-O bond indicated by the arrow in the figure below is formed.
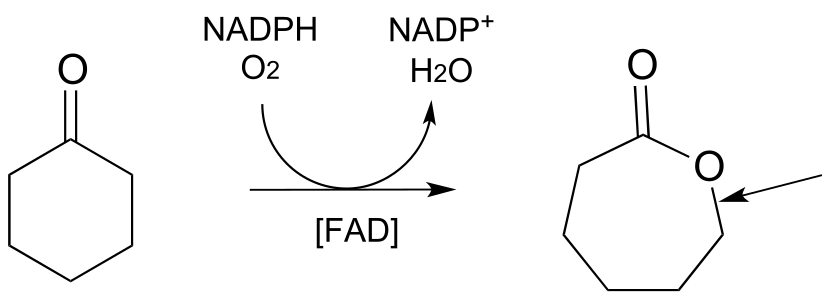
P15.11: Methanogens are a class of microorganisms in the domain archaea which inhabit a diversity of anaerobic (oxygen-lacking) environments, from the intestines of humans, to swamp mud, to the base of deep sea hot water vents. They obtain energy by reducing carbon dioxide to methane:
CO2 + 4H2 → CH4 + 2 H2O + energy
Methanogenesis’, like the oxidation of glucose in animals, is not accomplished in a single reaction - it requires a long series of enzymatic steps, and involves the participation of several unique coenzymes (if you are interested in learning more, see FEMS Microbiol. Rev. 1999, 23, 13 for a detailed review of the enzymatic reactions of methanogenesis).
The oxidation of methane to carbon dioxide when you burn natural gas for heating your house is obviously an exergonic process. How then is it possible that reducing carbon dioxide to methane could also be exergonic? Explain.
Multistep pathway prediction problems
In chapters 12 and 13, we were introduced to the challenge of mapping out potential multi-step transformations using the retrosynthesis approach, where we start with the more complex molecule and take it apart step be step. These problems have necessarily involved hypothetical, generalized transformations, because many of the reaction types involved in actual biochemical transformations were as-yet unfamiliar to us.
We have now reached a point in our study of organic reactivity where we can look at an actual metabolic pathway and probably recognize most of the reactions taking place - so were are ready to try our hand at mapping out real metabolic pathways.
Before we dive in, you may want to go back to the retrosynthesis interchapter and review the key elements of the retrosynthetic approach. You may (or may not) also need a little more practice with some simpler, shorter, hypothetical problems, similar to what we worked on in the last three chapters but incorporating some of the redox reactions that we have just finished learning about. These are provided in problems 15.12 and 15.13 below.
As before, your job is to draw out a ‘pathway diagram’ for each transformation, using the ‘arrow in - arrow out’ convention to indicate the role of other necessary participants in each reaction, such as ATP, NADH, water, or another organic molecule. An example for a simple two-step pathway is provided in problem 15.12 below. A three-step pathway would of course show two intermediate compounds.
Remember, it is most important that your proposed pathway be chemically reasonable - in other words, each hypothetical reaction that you propose should be very similar to a reaction pattern that we have seen in chapters 8-15. You should be able to put a name on each step: for example, ‘step 1 is a Claisen condensation; step 2 is a ketone reduction’, and so forth.
Also remember that there is usually no one correct way to approach problems like this - they are puzzles to solve, and success will be dependent in large part on having
a solid grasp on the chemical ‘tools’ available to us: in other words, the biochemical reaction types that we have been studying, starting with nucleophilic substitutions in chapter 8.
P15.12: hypothetical 2-step transformations:
Each of the generalized transformations below would be expected to require two enzymatic steps. Draw a reasonable pathway diagram for each transformation.
Example:
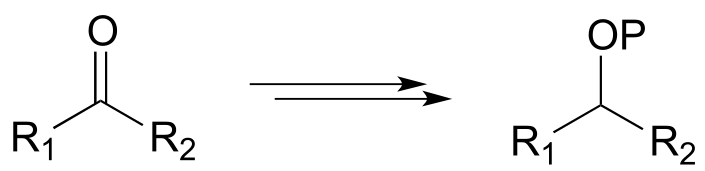
Diagram:
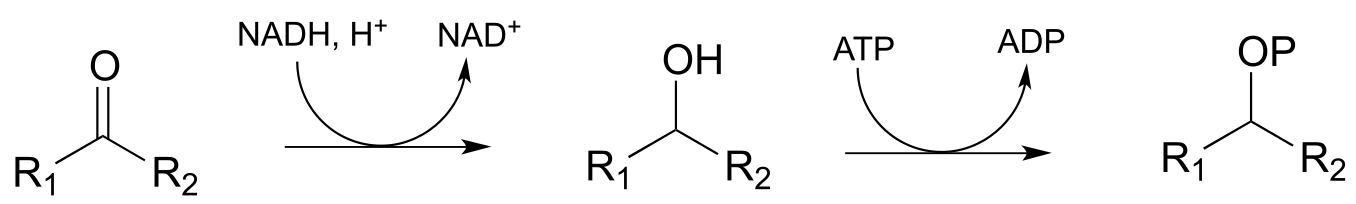
The first step is a nicotinamide-dependent ketone reduction/hydrogenation, and step 2 is ATP-dependent phosphorylation (ie. a kinase reaction). Note that the reducing agent in the first step could also be NADPH)
a)
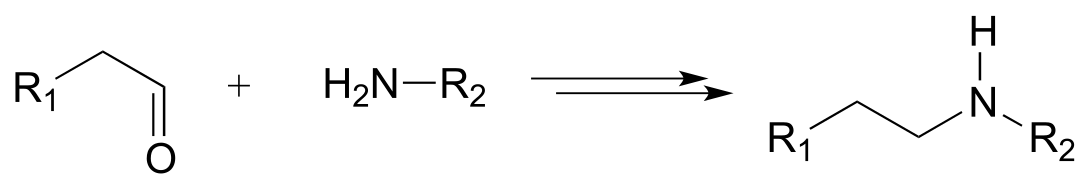
b)
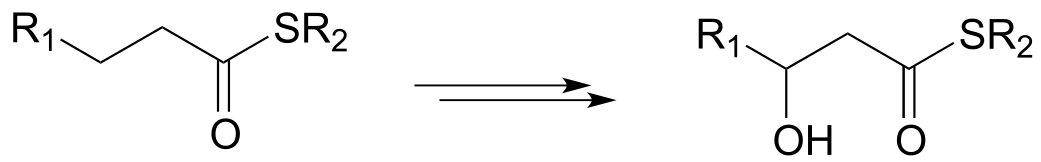
c)
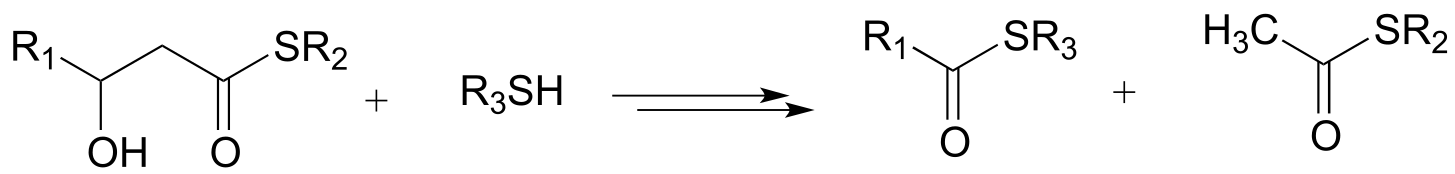
d)
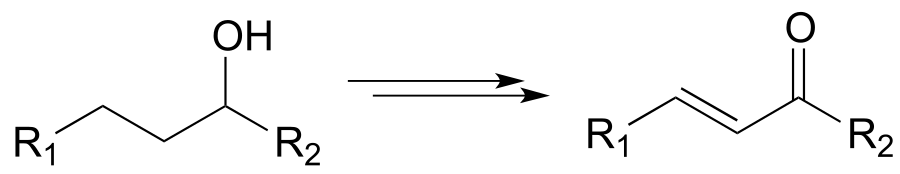
e)
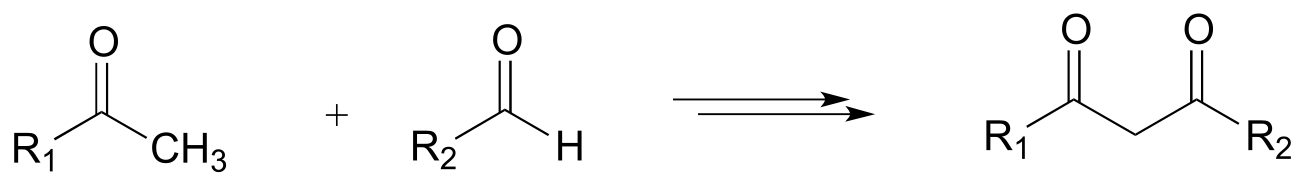
f)
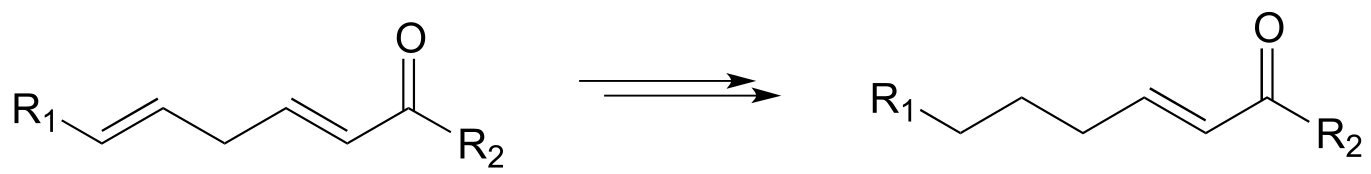
P15.13: hypothetical 3-step transformations:
Each of the generalized transformations below would be expected to require three enzymatic steps. Draw a reasonable pathway diagram for each transformation.
a)
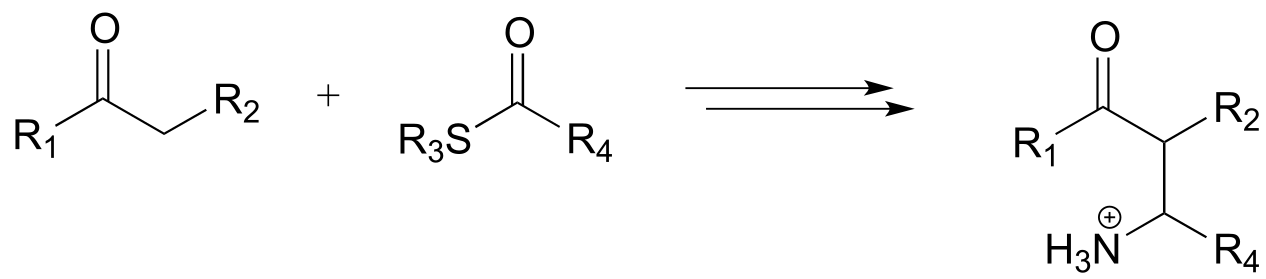
b)
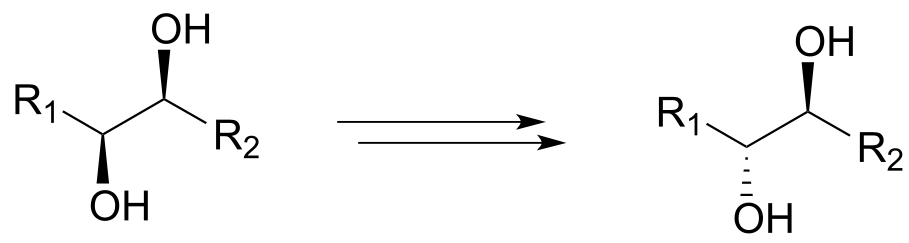
c)
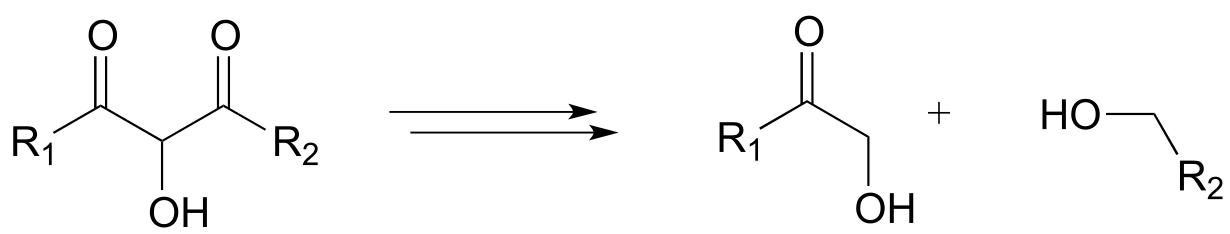
d)
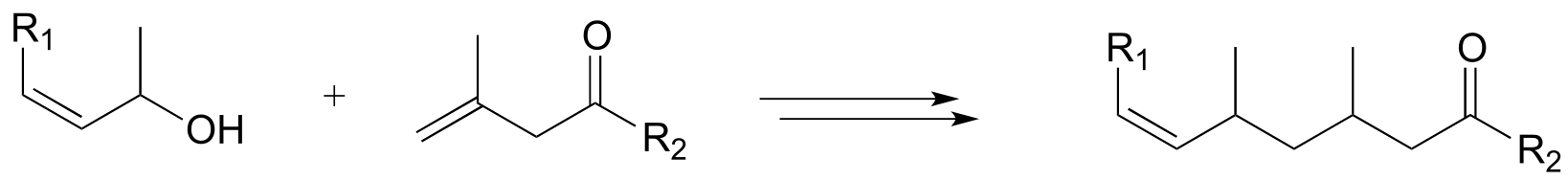
P15.14: Now, let’s try our hand at predicting the steps of some actual metabolic pathways. For each, draw a complete pathway diagram. As needed, use any of the coenzymes we have studied, water, and ammonia.
Note: you will probably find these quite challenging! Do not expect to be able to figure them out in a few minutes - rather, think of them as puzzles to work on over a period of time, sharing ideas and strategies with classmates. Remember, if one side of the transformation is larger or more complex, start there and work towards the simpler molecules. It is also a good idea, when applicable, to start the process by a) counting carbons on each side of the transformation, and b) identifying the key bond being formed (or broken) in the transformation.
a)
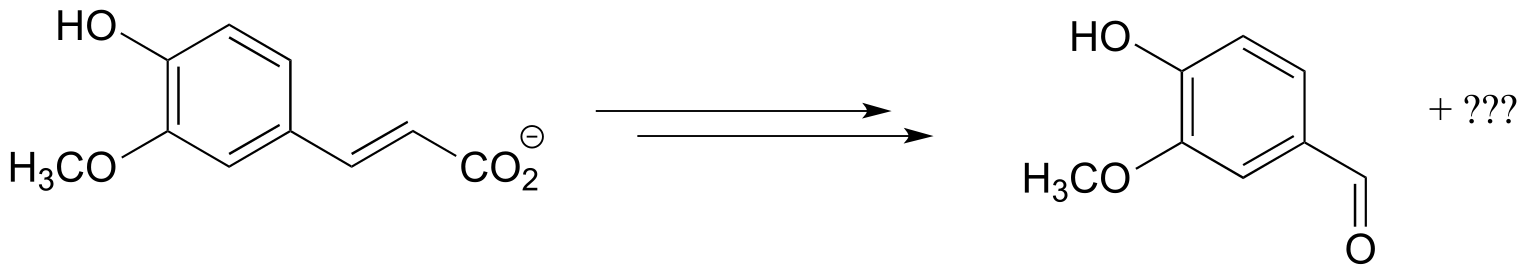
b)
c)

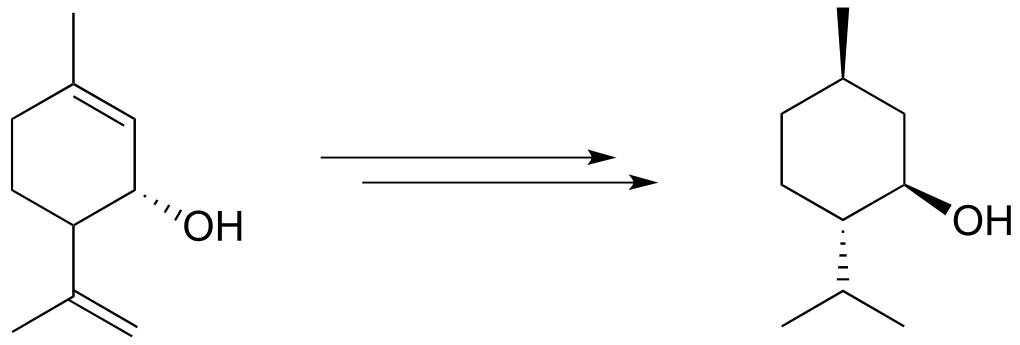
e)
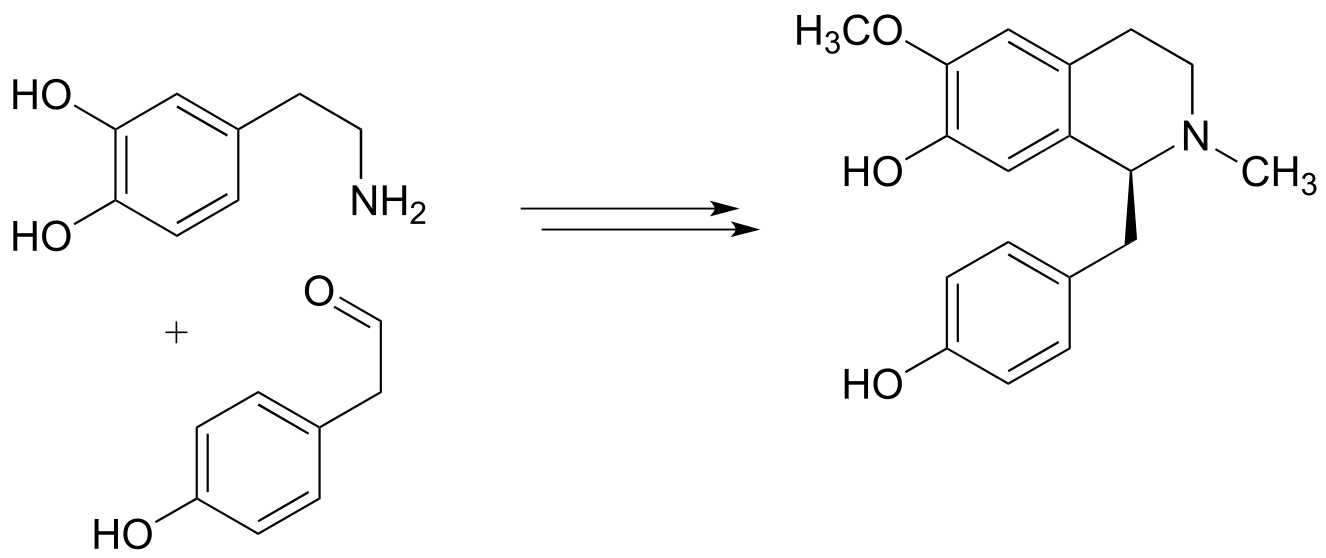
f)
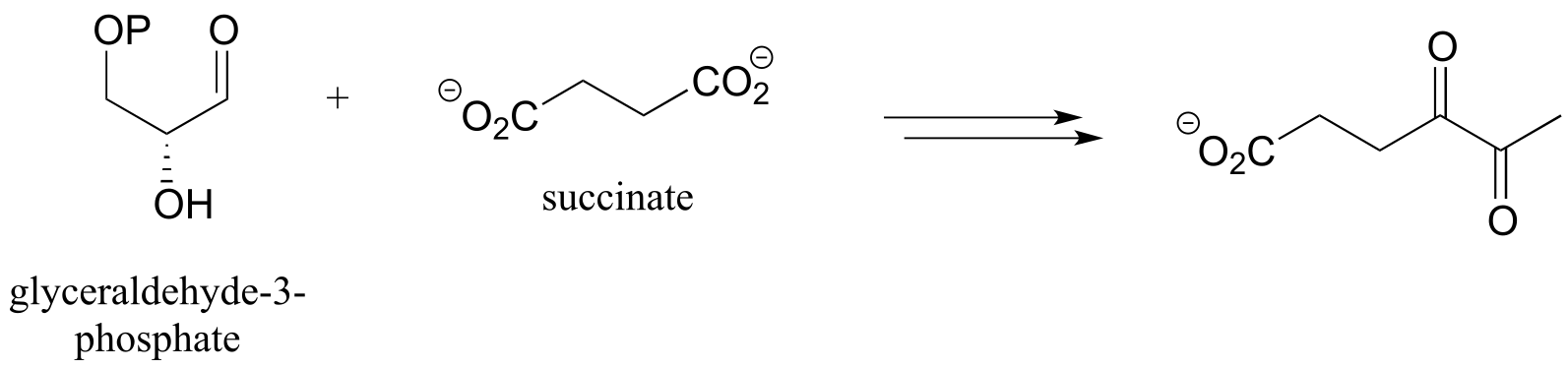
P15.15: Propose a pathway diagram for each of the metabolic pathways below. Note that some pathways contain steps that will be unfamiliar to you, and are therefore provided already.
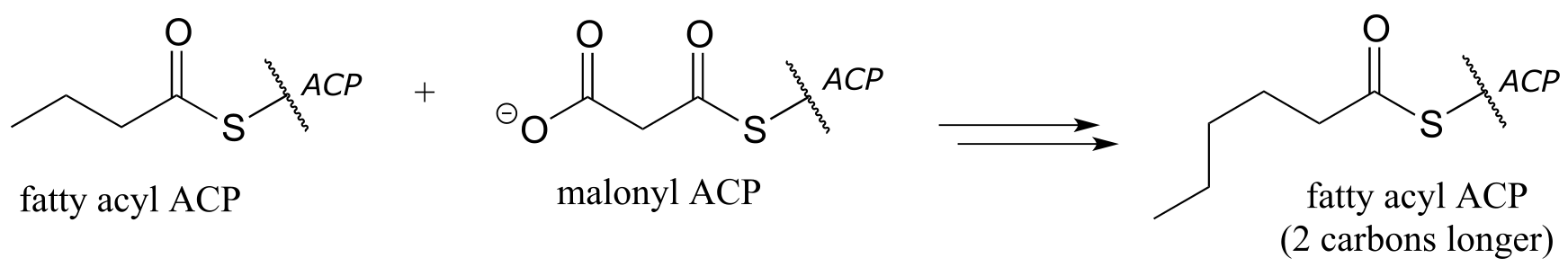
a) one cycle of fatty acid biosynthesis:
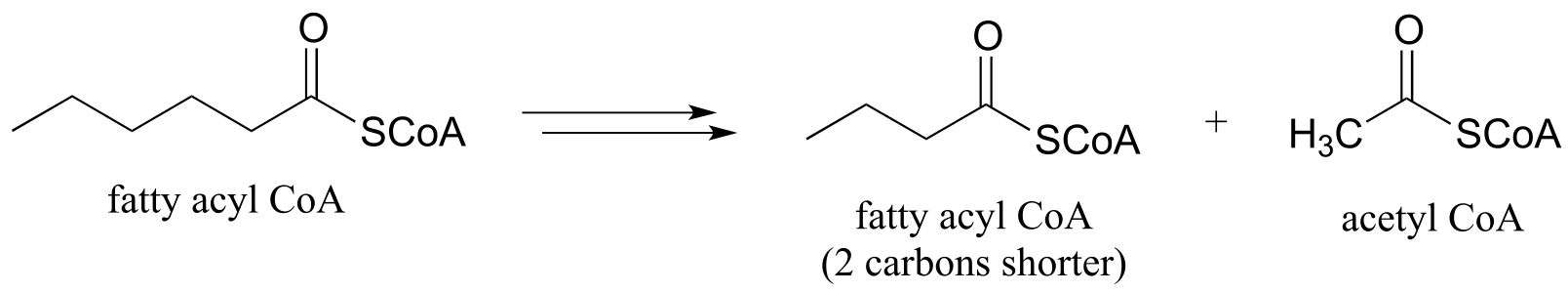
b) one cycle of fatty acid degradation:
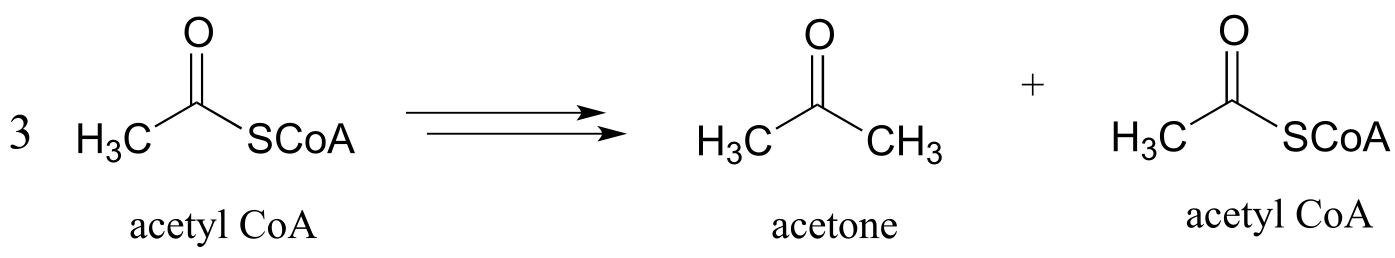
c) Diabetics and people who adopt an extreme low-carbohydrate diet sometimes have breath that smells like acetone, due to ‘ketone body’ formation that occurs when acetyl-CoA from fatty acid oxidation (see part (b) above) is not able to enter into the citric acid cycle. Draw a pathway diagram showing how three molecules of excess acetyl CoA combine to form acetone (all three acetyl-CoA molecules first link together, but one is left over at the end of the process).
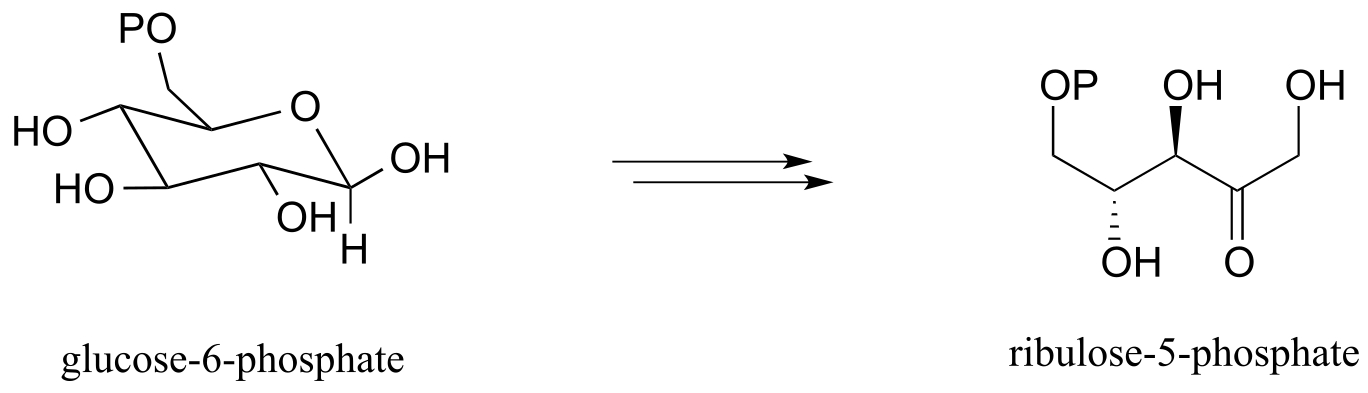
d) Pentose phosphate pathway (oxidative branch):
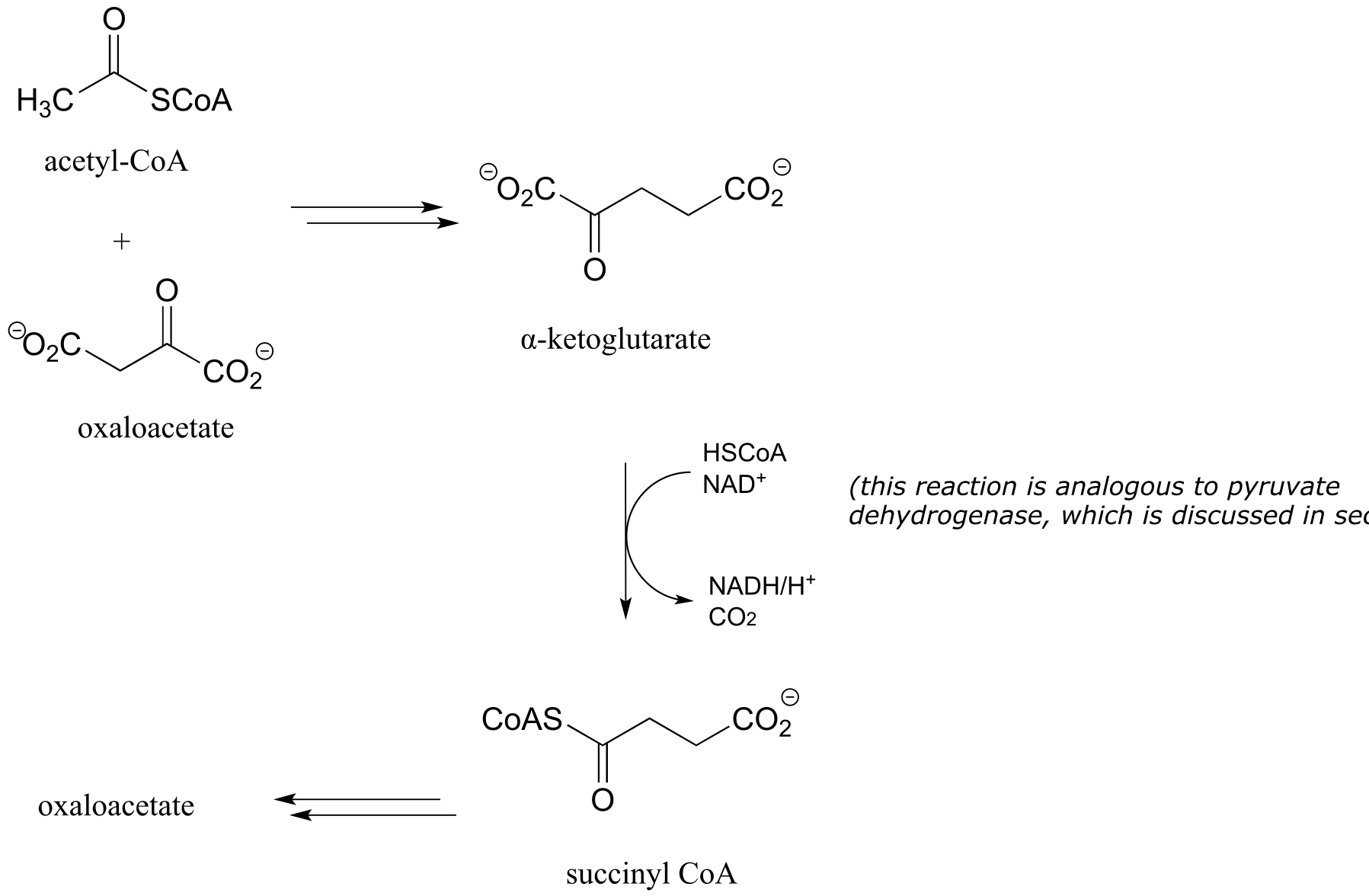
e) Citric acid (Krebs) cycle:
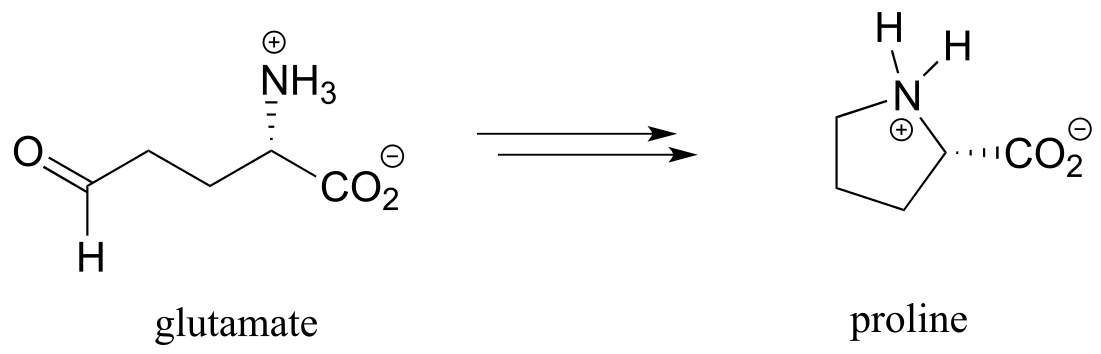
f) proline biosynthesis:
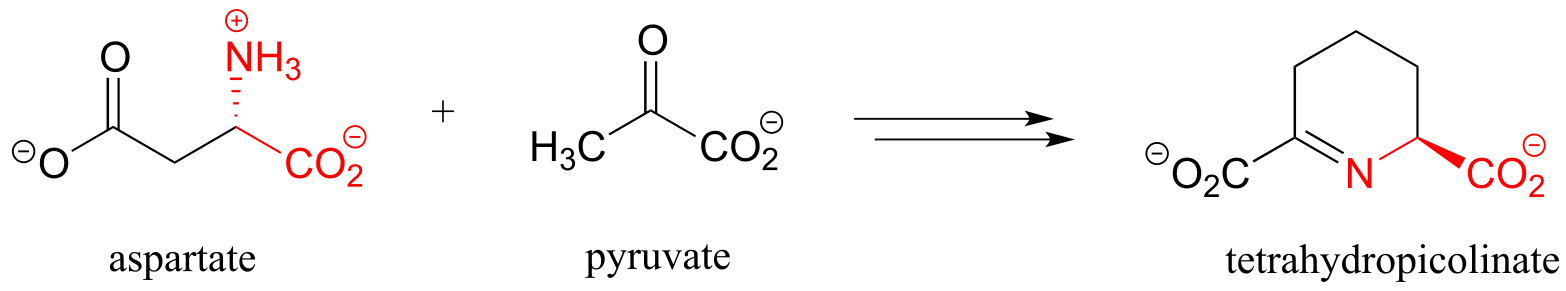
g) First half of lysine biosynthesis:
h) From the biosynthesis of membrane lipid in archaea: J. Bacteriol. 2003, 185, 1181