17: Coenzymes
Contents
17: Coenzymes#

Photo: Taconis Kryn, Library and Archives Canada - PA169941
The old black and white photograph is haunting. A young boy, perhaps 10 or 11 years old, huddles against a wall outside a soup kitchen, his mouth in an odd twisted shape that could be expressing either pain or defiance, his eyes staring straight into those of the viewer. Tucked into his pants, almost like a pistol in a holster, is a metal spoon.
The photograph was taken in the Netherlands in 1945, at the height of what the Dutch people still refer to simply as “The Hunger Winter”. With the western part of the country still occupied by the Nazis, the Dutch resistance government, based in London, had called for a railway strike with the aim of stopping German troop movements before a planned airborne invasion by Allied forces. In retaliation, the Germans cut off all food shipments to cities in the western Netherlands. The Allied invasion failed to liberate the country, and the winter of 1944-1945 turned out to be bitterly cold. With food supplies dwindling, rations were cut first to 1000 calories per day, then to 500. People resorted to eating grass and tulip bulbs just to stay alive. Over 20,000 people died of starvation before food shipments were restored in the spring.
As tragic as the Hunger Winter was for the Dutch people, some good did come from it. For medical scientists, the event became a unique ‘social experiment’: unlike most episodes of famine throughout human history, the Hunger Winter had a clearly defined beginning, end, and geographic boundary, and it occurred in a technologically advanced society that continued to keep thorough records before, during, and after the ordeal. Scientists knew exactly who suffered from famine and for precisely how long, and in the years that followed they were able to look at the long-term effects of famine, particularly on developing embryos. Researchers found that babies who had been conceived during the famine were born with a significantly higher incidence of neurological birth defects such as spina bifida, a condition in which a portion of the neural tube protrudes from between vertebrae which did not properly fuse together during fetal development. Later in life, members of this same cohort of ‘famine babies’ were more likely to be obese, and to suffer from schizophrenia.
These initial findings spurred interest in further research into the consequences of prenatal deprivation. In particular, carefully controlled studies later led to the recognition of the importance of the vitamin folate in ensuring proper neurological development in early-term fetuses.
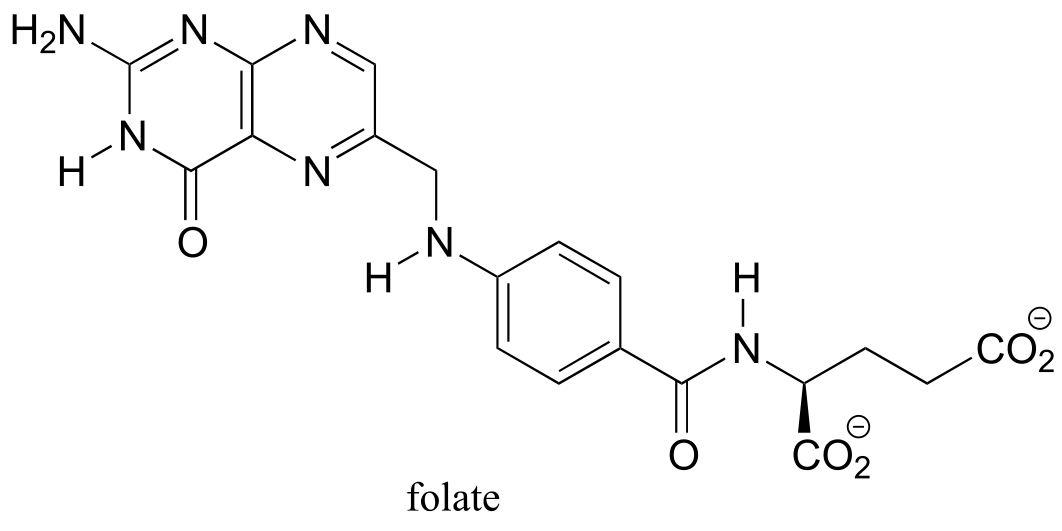
fig 1a
Folate - the conjugate base of folic acid - is an organic coenzyme: a helper molecule that binds in the active site of certain enzymes and plays a critical role in the biochemical reaction being catalyzed. Recall that we have seen coenzymes at work before: SAM, ATP, NAD(P)+ and NAD(P)H, flavin and glutathione are all important coenzymes with which we are already familiar.
Because prenatal folate deficiency was found to be directly related to the incidence of spina bifida and other neural tube defects, health officials in the United States and many other countries changed their official guidelines to include a specific recommendation that women begin taking folate supplements as soon as they knew that they are pregnant, or better yet as soon as they begin trying to become pregnant. A number of studies conducted during the 1980s and early 1990s consistently showed that folate supplementation correlated with a 50-70% reduction in neural tube defects.
The molecular role of folate in prenatal neurological development is not understood in detail, but most researchers agree that it probably has a lot to do with DNA biosynthesis. Like *S-*adenosyl methionine (SAM), folate functions in 1-carbon transfer reactions, including several critical steps in the nucleic acid biosynthesis pathways. The rapidly dividing cells of the developing brain of an early term fetus appear to be especially sensitive to folate deficiency in the mother’s diet: insufficient folate leads to impaired DNA biosynthesis, which in turn leads to defects in brain development.
Folate also serves as a 1-carbon donor in the pathway by which SAM is regenerated after it donates a methyl group. You may recall from the introduction to chapter 8 that methylation of cytosine bases in DNA by SAM results in permanent changes to a individual’s genome - this was the reason why the two ‘identical’ twin sisters in that introductory story turned out to be, as they grew older, not so identical after all. It is likely that the folate deprivation that afflicted expectant mothers during the Dutch Hunger Winter also caused epigenetic changes (in other words, changes in the extent of DNA methylation) in their developing fetuses, which decades later manifested in the form of an increased incidence of conditions such as obesity and schizophrenia. All the more reason, we now know, to make sure that women get plenty of folate in their diet early in the first trimester of pregnancy.
In this final chapter, we focus on the organic chemistry of folate, along with three other coenzymes: pyridoxal phosphate, thiamine diphosphate, and lipoamide. Humans can synthesize lipoamide, but we depend on dietary sources for the other three: pyridoxal phosphate is a form of vitamin B6, and thiamine diphosphate is a form of vitamin B1. In a mechanistic sense, there is really nothing new in this chapter. All of the reaction mechanism types that we will see are already familiar to us, ranging from nucleophilic substitutions (chapter 8) to disulfide exchanges (chapter 15). We will soon see, however, how each coenzyme plays its own specific and crucial role in assisting enzymes with the catalysis of key reactions of metabolism. We will begin with pyridoxal phosphate and its various roles in amino acid metabolism.
Additional reading:
http://www.naturalhistorymag.com/features/142195/beyond-dna-epigenetics
Section 17.1: Pyridoxal phosphate (Vitamin B6)#
The coenzyme pyridoxal phosphate (commonly abbreviated PLP) is the active form of vitamin B6, or pyridoxine.
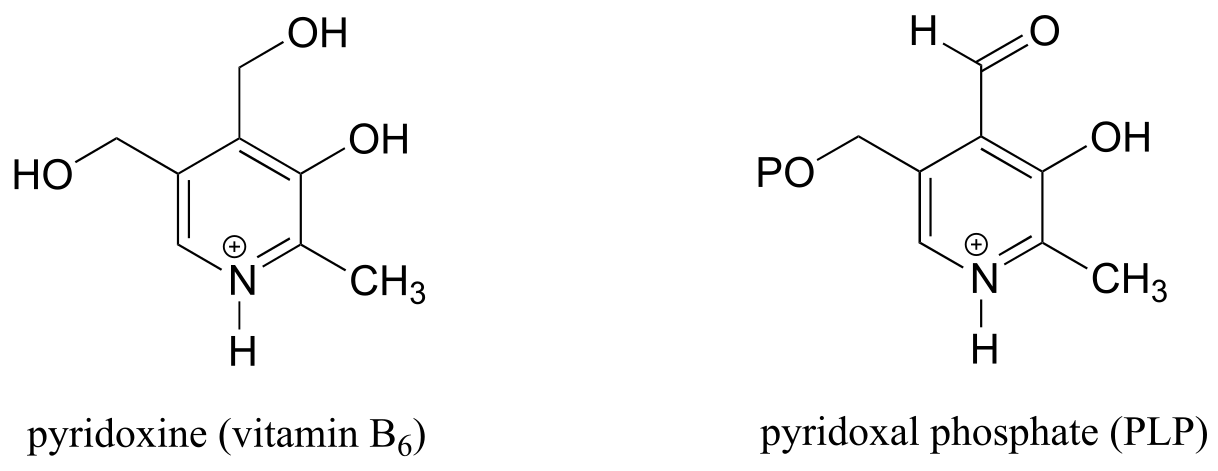
fig 1
PLP is required for over 100 different reactions in human metabolism, primarily in the various amino acid biosynthetic and degradation pathways. The essential function of PLP is to act as an ‘electron sink’, stabilizing a negative formal charge that develops on key reaction intermediates. Some of reactions will be familiar to you from chapter 12 and 13: we will see examples, for instance, of PLP-dependent α-carbon racemization, as well as aldol- and Claisen-type reactions. Other reactions will be less familiar: for example, the participation of PLP allows for decarboxylation of amino acids, a chemical step which would be highly unlikely without the coenzyme, and PLP is also required for a very important class of biochemical transformation called ‘transamination’, in which the amino group of an amino acid is transferred to an acceptor molecule. Before we dive into the reactions themselves, though, we need to begin by looking at a key preliminary step that is common to all of the PLP reactions we will see in this section.
17.1A: PLP in the active site: the imine linkage#
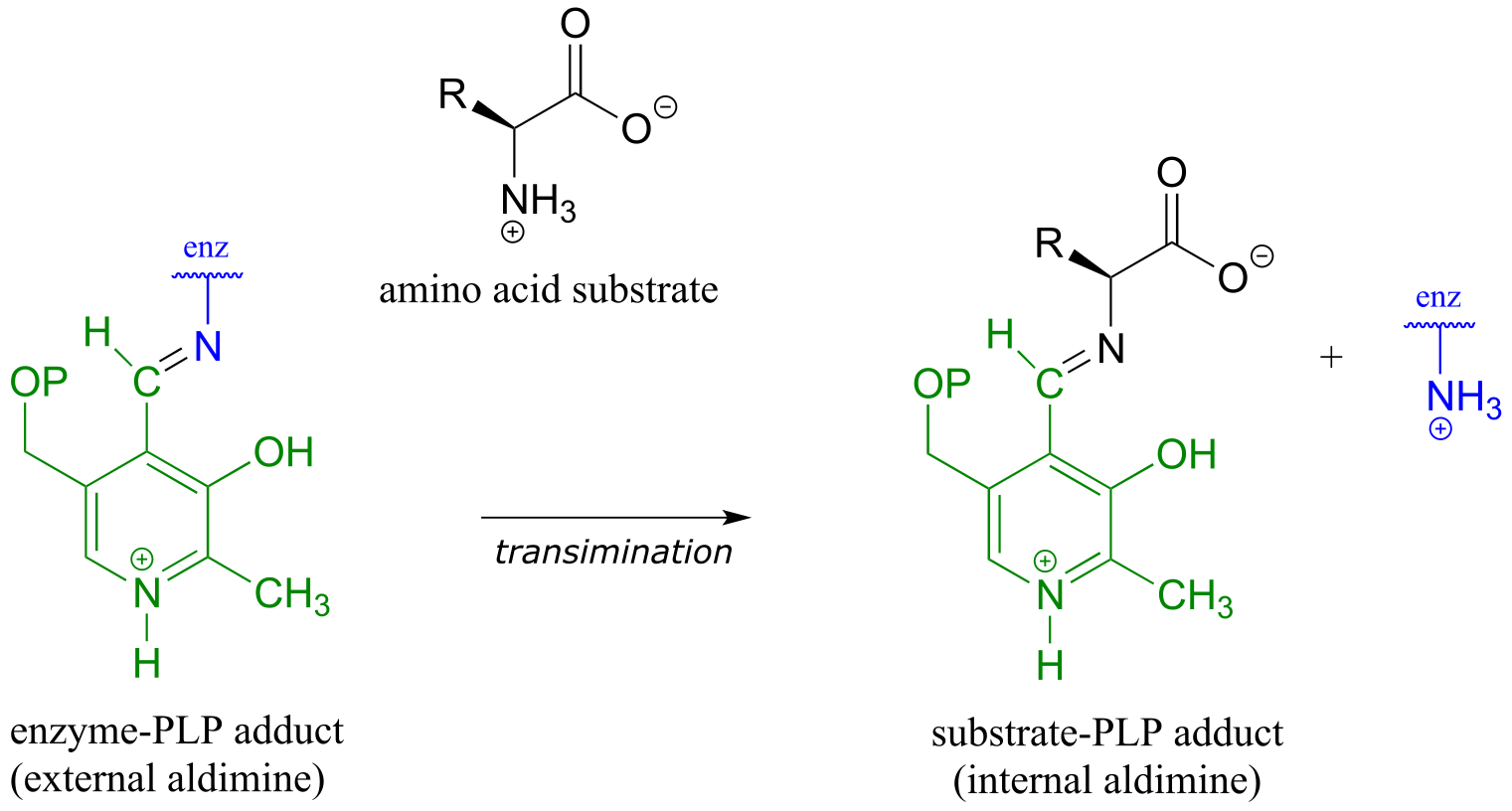
The common catalytic cycle of a PLP-dependent enzyme begins and ends with the coenzyme covalently linked to the enzyme’s active site through an imine linkage between the aldehyde carbon of PLP and the amine group of a lysine residue (see section 10.5 to review the mechanism for imine formation). For a PLP-dependent enzyme to become active, a PLP molecule must first enter the active site of an enzyme and form an imine link to the lysine. This state is often referred to as an external aldimine.
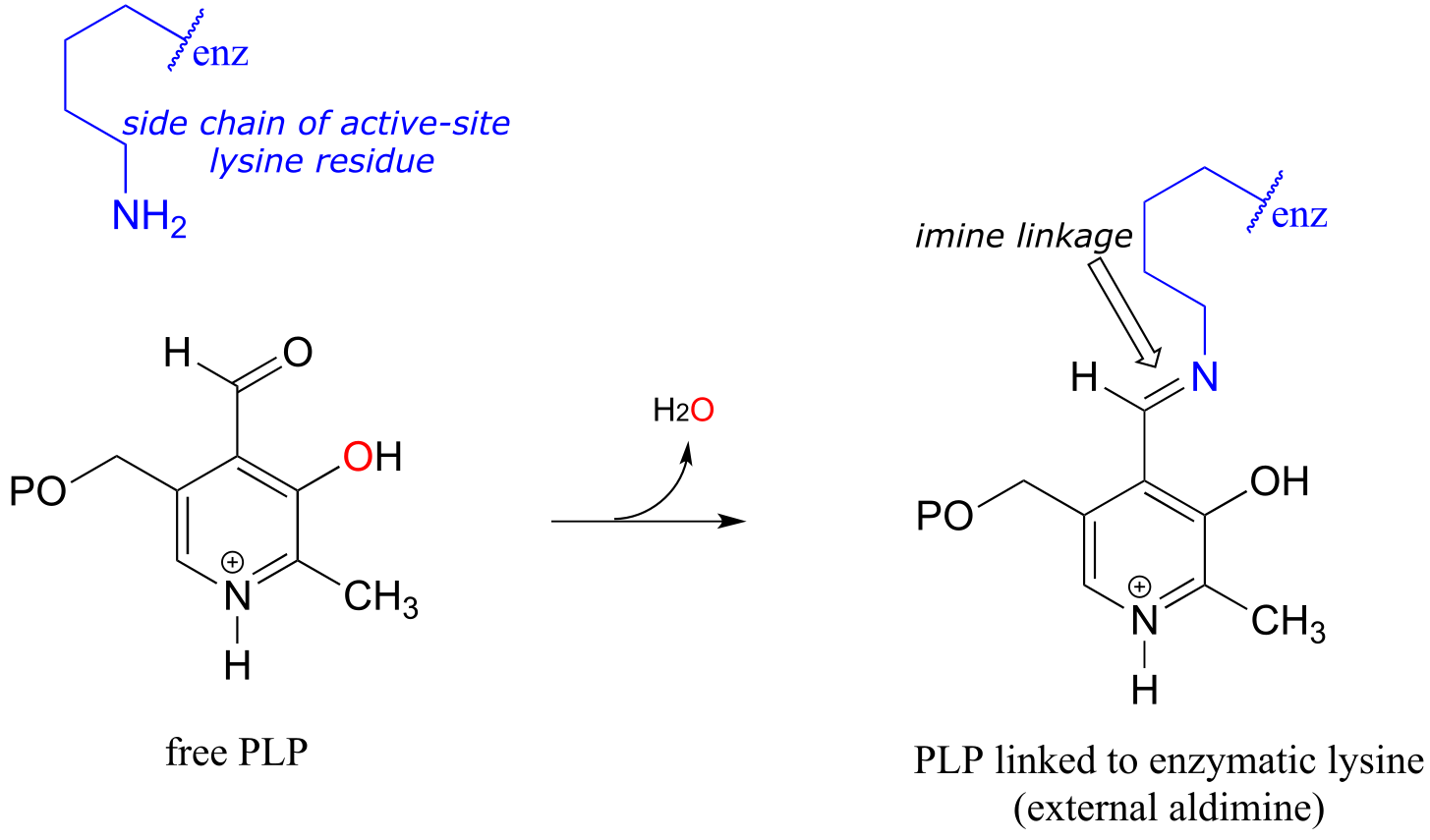
fig 2
The first step of virtually all PLP-dependent reactions is transimination (section 10.5), as the amino group on the amino acid substrate displaces the amino group of the enzymatic lysine. This state - where the coenzyme is covalently linked to the substrate or product of the reaction - is often referred to as an internal aldimine.
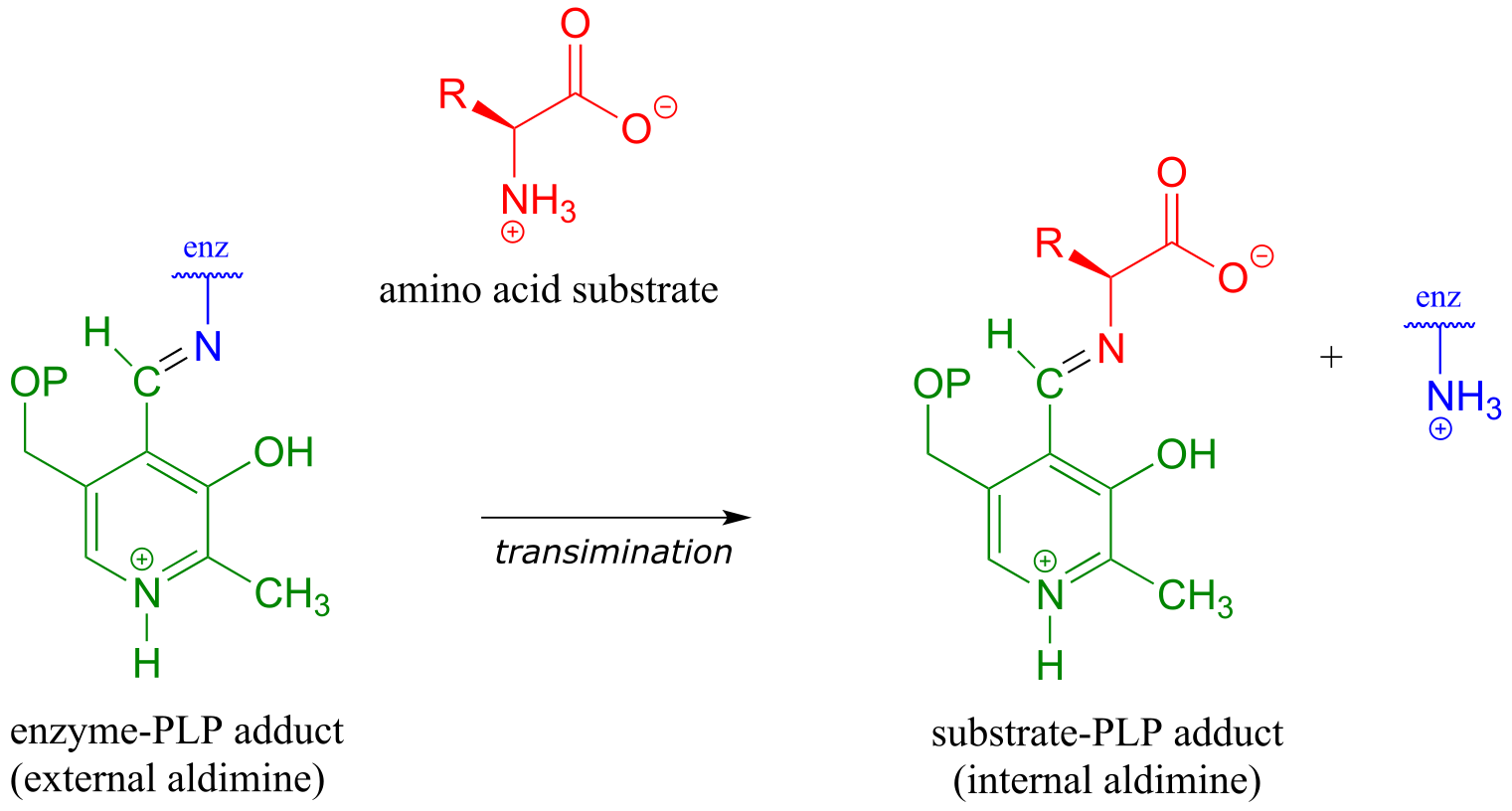
fig 3
With the preliminary transimination accomplished, the real PLP chemistry is ready to start. The versatility of PLP in terms of its ability to assist with a wide variety of reaction types is illustrated by the figure below, showing how, depending upon the reaction/enzyme in question, PLP can assist in the cleavage of any one of the four bonds to the α-carbon of the amino acid substrate.
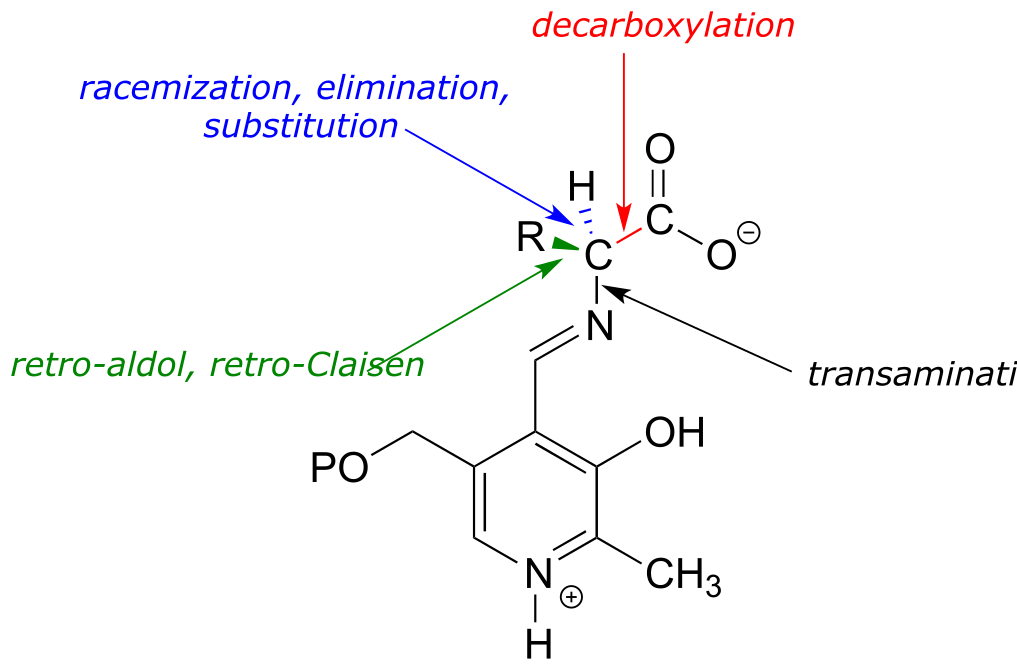
fig 4
Let’s look first at the reaction catalyzed by PLP-dependent alanine racemase. (EC 5.1.1.1).
17.1B: PLP-dependent amino acid racemization#
In section 12.2B we saw an example of a PLP-independent amino acid racemization reaction, in which the negatively-charged intermediate was simply the enolate form of a carboxylate:
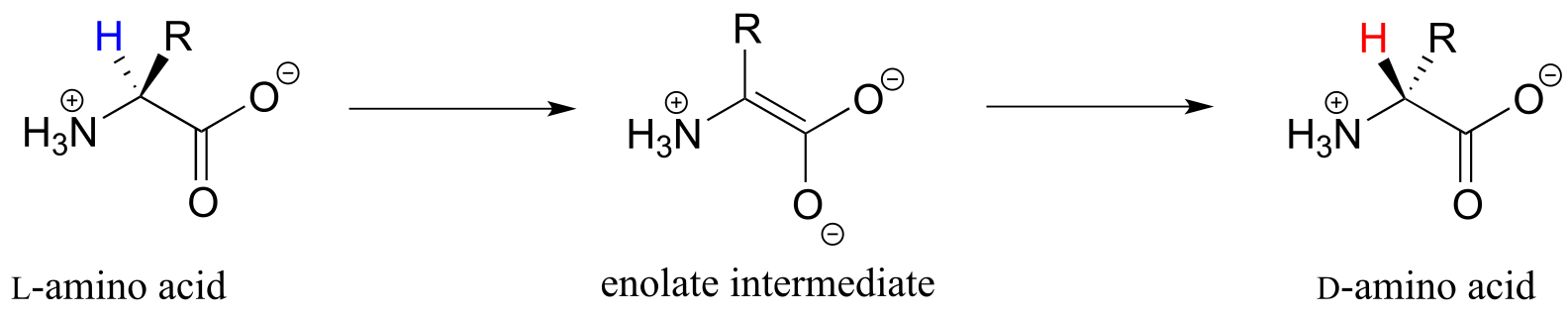
fig 5
Many other amino acid racemase reactions, however, require the participation of PLP.
Like all other PLP-dependent reactions that we will see in this section, PLP-dependent amino acid racemization begins with a preliminary step in which the substrate becomes attached to the coenzyme through a transimination. Once it is linked to PLP in the active site, the α-proton of an amino acid substrate is abstracted by an active site base (step 1 below). The negative charge on the carbanion intermediate can, of course, be delocalized to the carboxylate group. The PLP coenzyme, however, provides an expanded network of conjugated π-bonds over which the electron density can be delocalized all the way down to the PLP nitrogen. This is what we mean when we say that the job of PLP is to act as an ‘electron sink’: the coenzyme is very efficient at absorbing, or delocalizing, the excess electron density on the deprotonated α-carbon of the reaction intermediate. PLP is helping the enzyme to increase the acidity of the α-hydrogen by stabilizing the conjugate base. A PLP-stabilized carbanion intermediate is commonly referred to as a quinonoid intermediate. Note that in the overall reaction equation below, PLP appears below the reaction arrow in brackets, indicating that it participates in the mechanism but is regenerated as part of the reaction cycle.
PLP-dependent amino acid racemization:
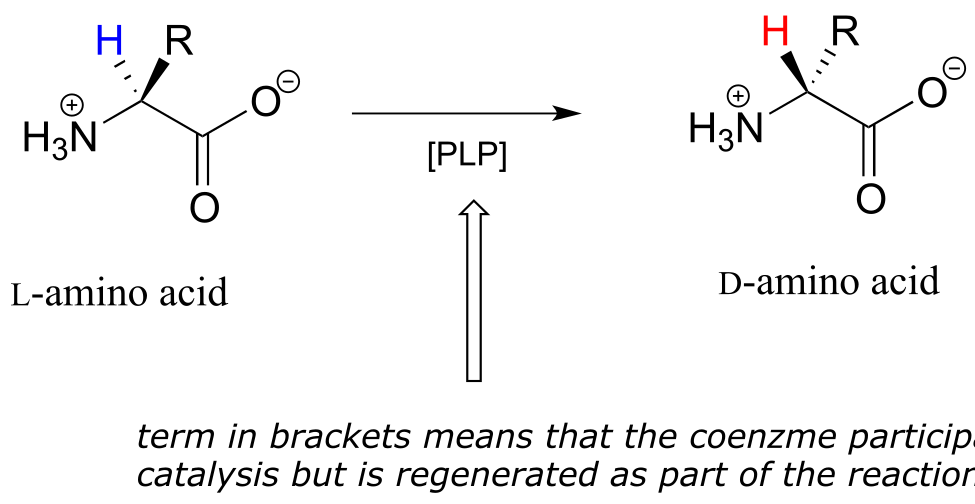
Mechanism:
Preliminary step - transimination:
First step - deprotonation:
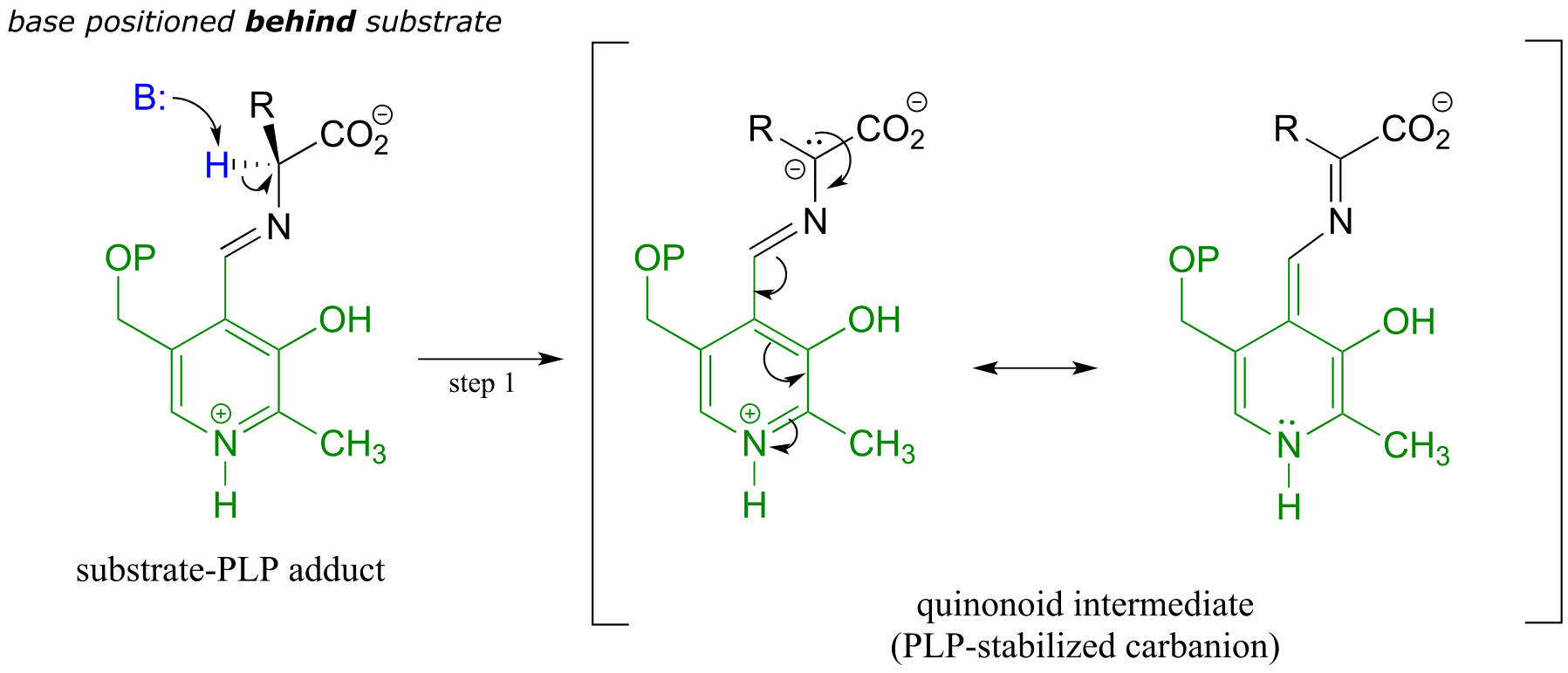
Second step - reprotonation from the opposite side:
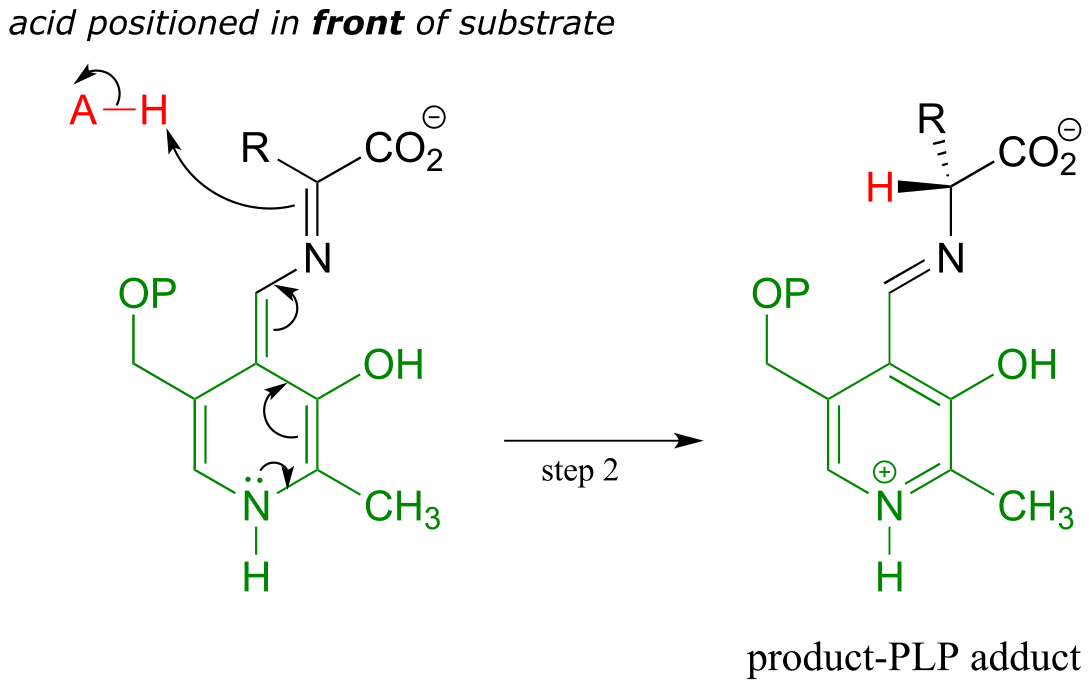
Final step - transimination:
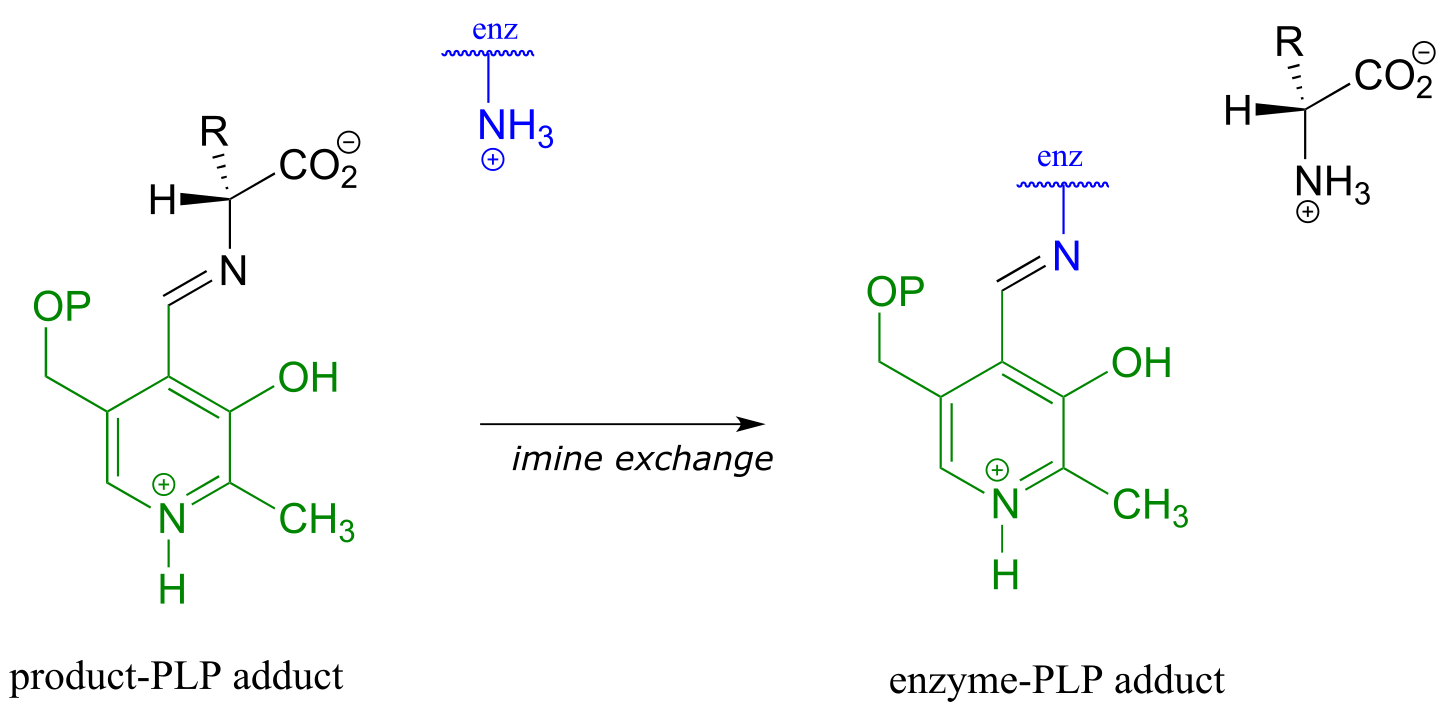
fig 7
fig 6 fig 6a
Just as in the PLP-independent racemase reactions, reprotonation occurs on the opposite side of the substrate (step 2), leading to the D-amino acid product.
All that remains is the final imine exchange which frees the D-amino acid product and re-attaches the coenzyme to the enzymatic lysine side-chain, ready to begin another catalytic cycle.
To simplify matters, from here on we will not include the preliminary and final transimination steps in our PLP reaction figures - we will only show mechanistic steps that occur while the substrate is attached to the coenzyme (the internal aldimine forms).
17.1C: PLP-dependent decarboxylation#
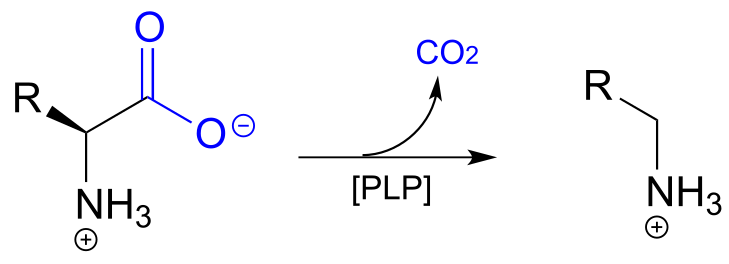
In the amino acid racemase reaction above, PLP assisted in breaking the α-carbon to α-proton bond of the amino acid. Other PLP-dependent enzymes can catalyze the breaking of the bond between the α-carbon and the carboxylate carbon by stabilizing the resulting carbanion intermediate: these are simply decarboxylation reactions.
PLP-depended amino acid decarboxylation:
Mechanism:
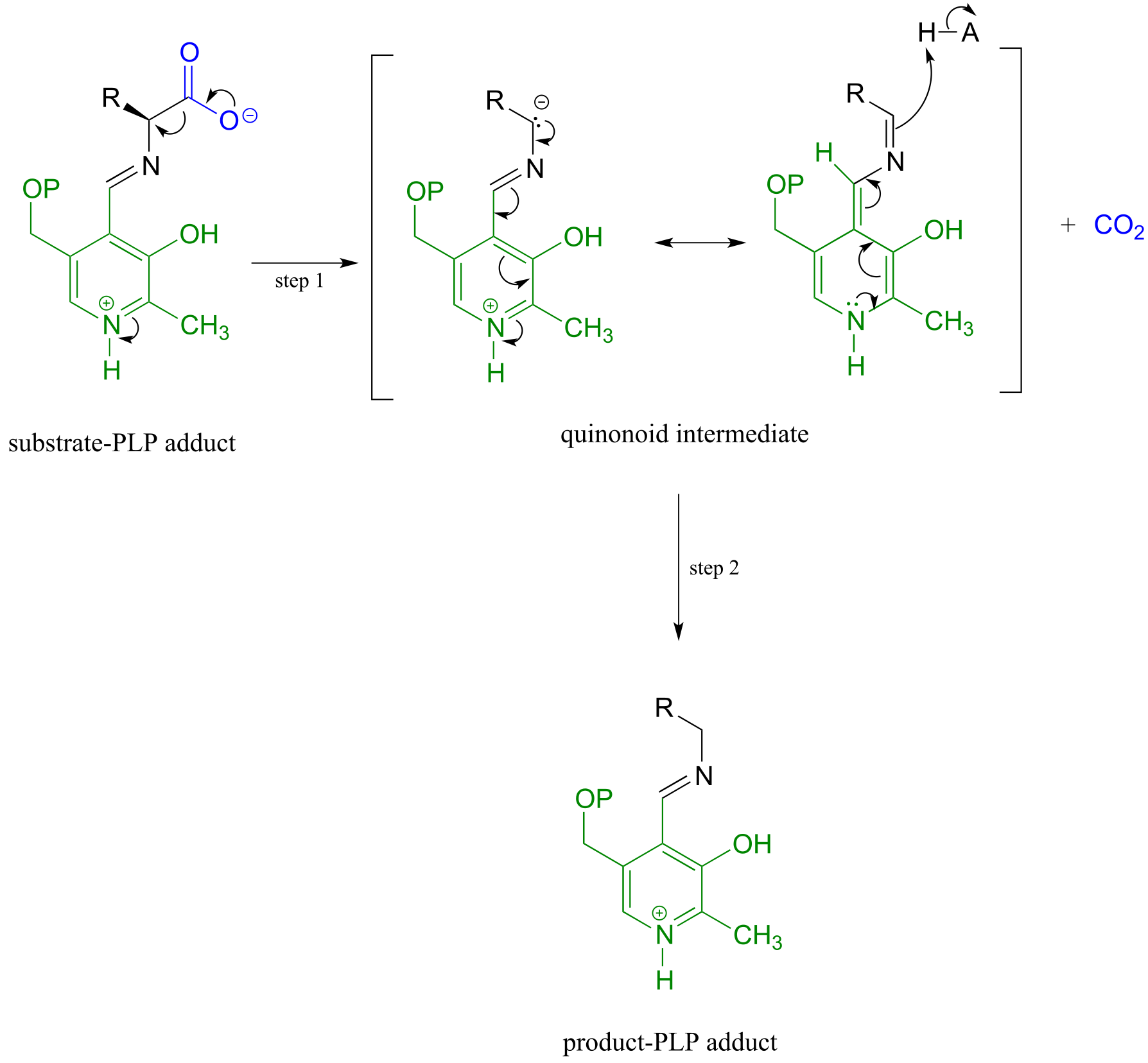
fig 8 fig 9
Notice something very important here: while in racemization reactions the assistance of PLP can be seen as ‘optional’ (in the sense that some racemase enzyme use PLP and others do not), the coenzyme is essential for amino acid decarboxylation steps. Without PLP, there is no way to stabilize the carbanion intermediate, and decarboxylation is not a chemically reasonable step.
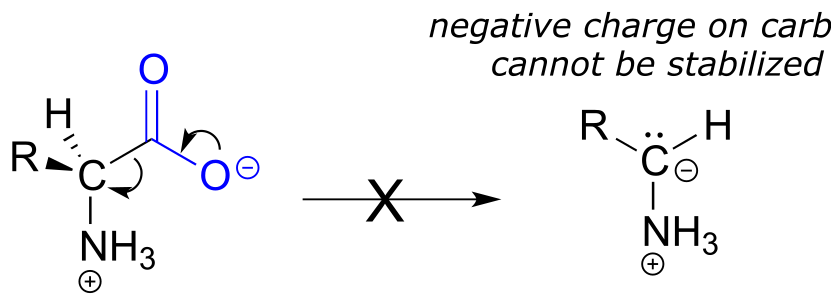
fig 10
One example of a PLP-facilitated decarboxylation reaction is the final step in the lysine biosynthesis pathway: (EC 4.1.1.20).
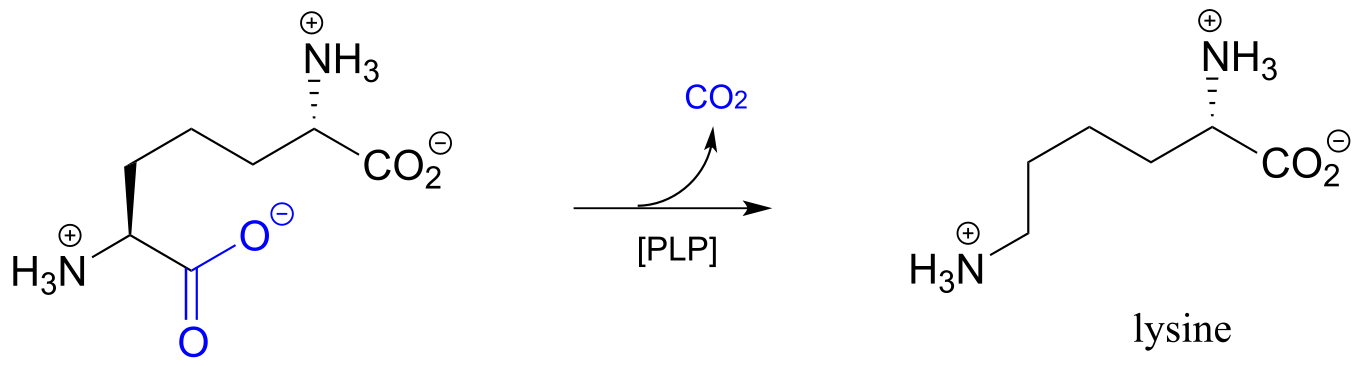
fig 11
Exercise 17.1: Draw mechanistic arrows for the carbon-carbon bond-breaking step of the PLP-dependent decarboxylation reaction above.
17.1D: PLP-dependent retroaldol and retro-Claisen cleavage#
(It would be a good idea before reading this section to review aldol/retro-aldol and Claisen/retro-Claisen reaction mechanisms in sections 12.3 and 13.3, respectively)
So far we have seen PLP playing a role in breaking the bond between the α-carbon and its α-proton (in the racemization reaction), and the bond between the α-carbon and carboxylate carbon (in the decarboxylation reaction).
fig 4
Other PLP-dependent enzymes catalyze cleavage of the bond between the α-carbon and the first carbon on the amino acid side chain, otherwise known as the β-carbon. In the serine degradation pathway, serine is first converted to glycine by a retro-aldol cleavage reaction. Although a reasonable mechanism could be proposed without the participation of PLP, this reaction in fact requires the coenzyme to assist in stabilization of the negative charge on the carbanion intermediate.
A PLP-dependent retro-aldol cleavage reaction
(serine hydroxymethyl transferase, EC 2.1.2.1)
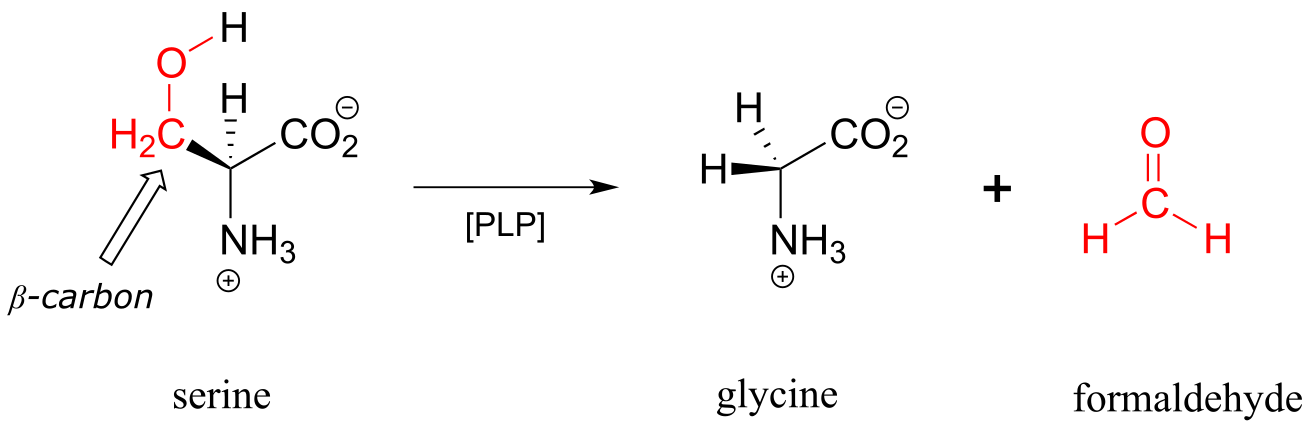
Mechanism:
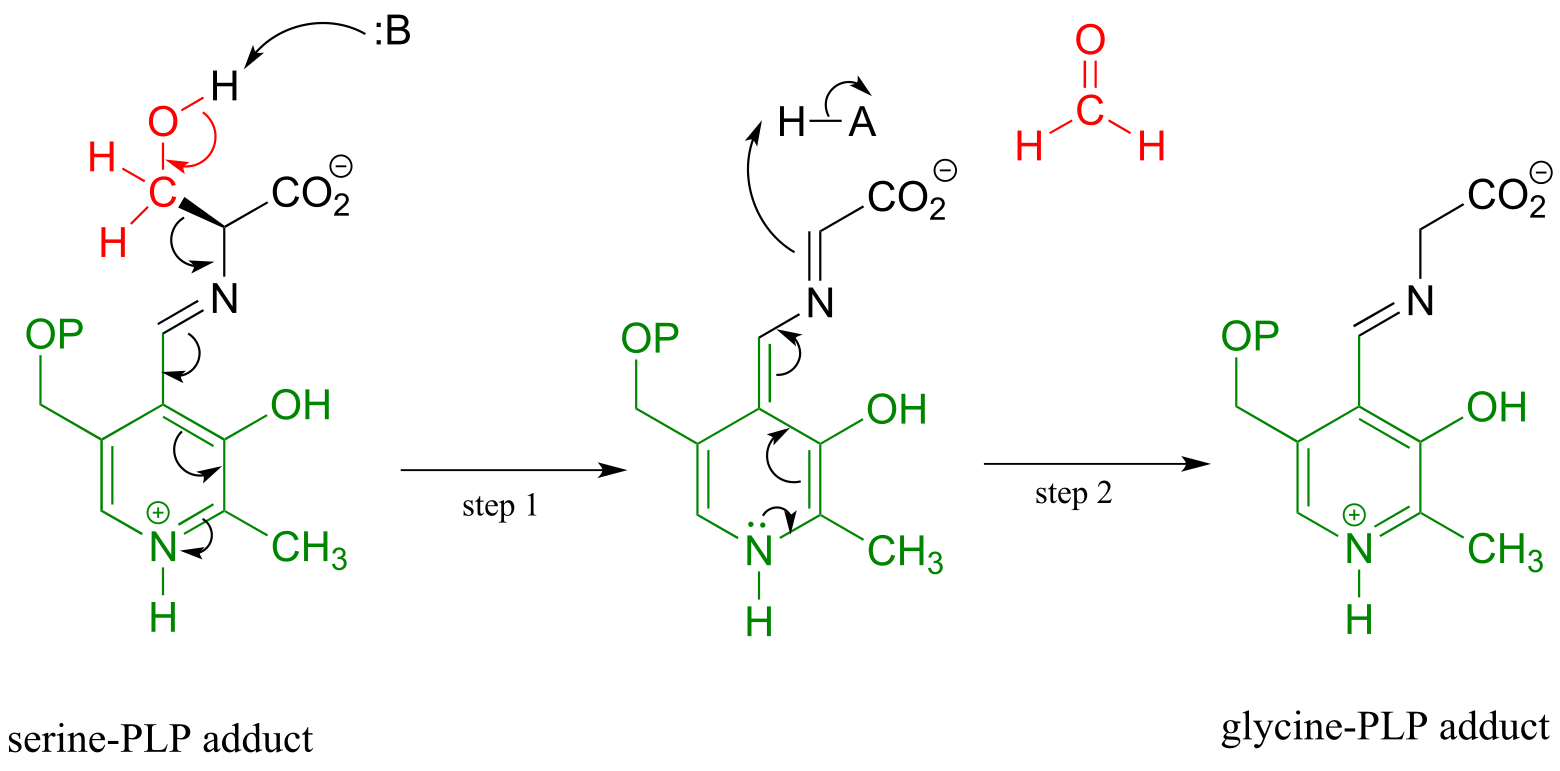
fig 12 fig 13
Note that, in this reaction just as in the racemase reaction described previously, the key intermediate is a PLP-stabilized carbanion, or quinonoid.
What happens to the (toxic!) formaldehyde produced in this reaction? We will see later in this chapter how the serine hydroxymethyltransferase enzyme goes on to use another coenzyme called tetrahydrofolate to prevent the formaldehyde from leaving the active site and causing damage to the cell.
PLP also assists in retro-Claisen cleavage reactions (section 13.3C), such as this step in the degradation of threonine. (EC 2.3.1.29)
A PLP-dependent retro-Claisen reaction:

Mechanism:
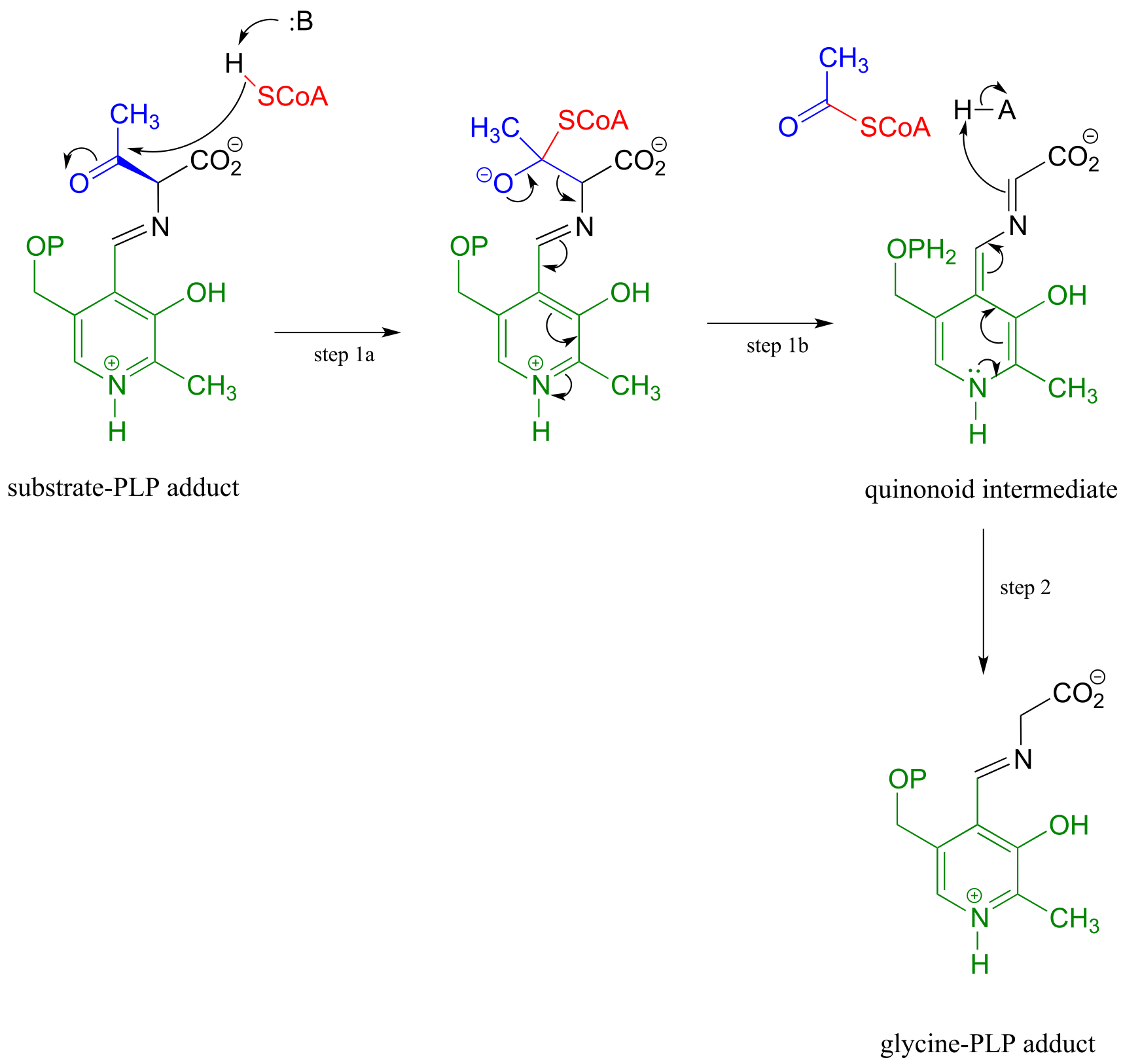
fig 14 fig 15
Notice how, like the retro-aldol reaction, the bond between the α-carbon and the β-carbon of the amino acid substrate is broken (in step 1b).
17.1-E: PLP-dependent transamination#
One of the most important reaction types in amino acid metabolism is transamination, in which an amino group on a donor molecule (often an amino acid) is transferred to a ketone or aldehyde acceptor molecule.
A transamination reaction:
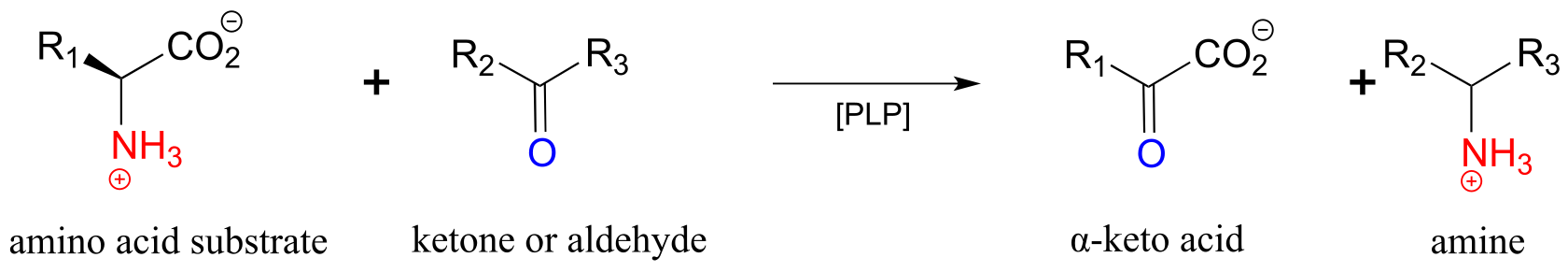
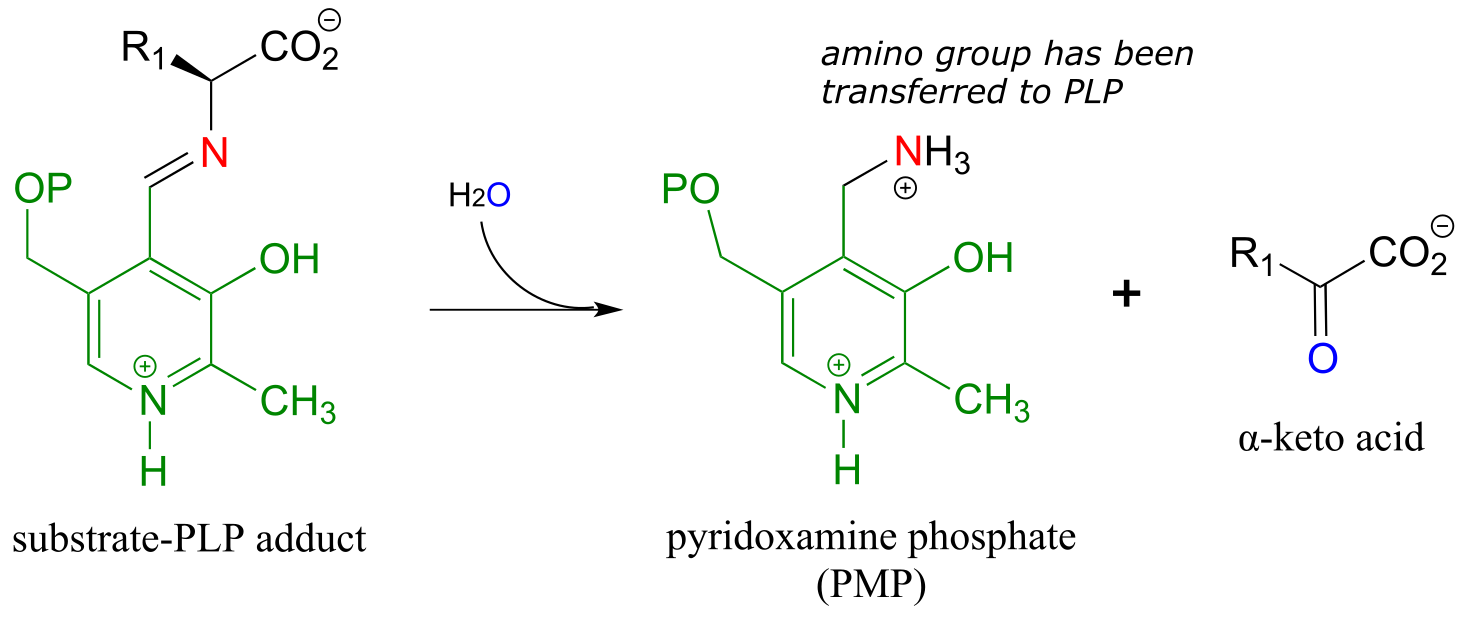
Transamination phase 1 (transfer of amino group from amino acid substrate to coenzyme)
Mechanism:
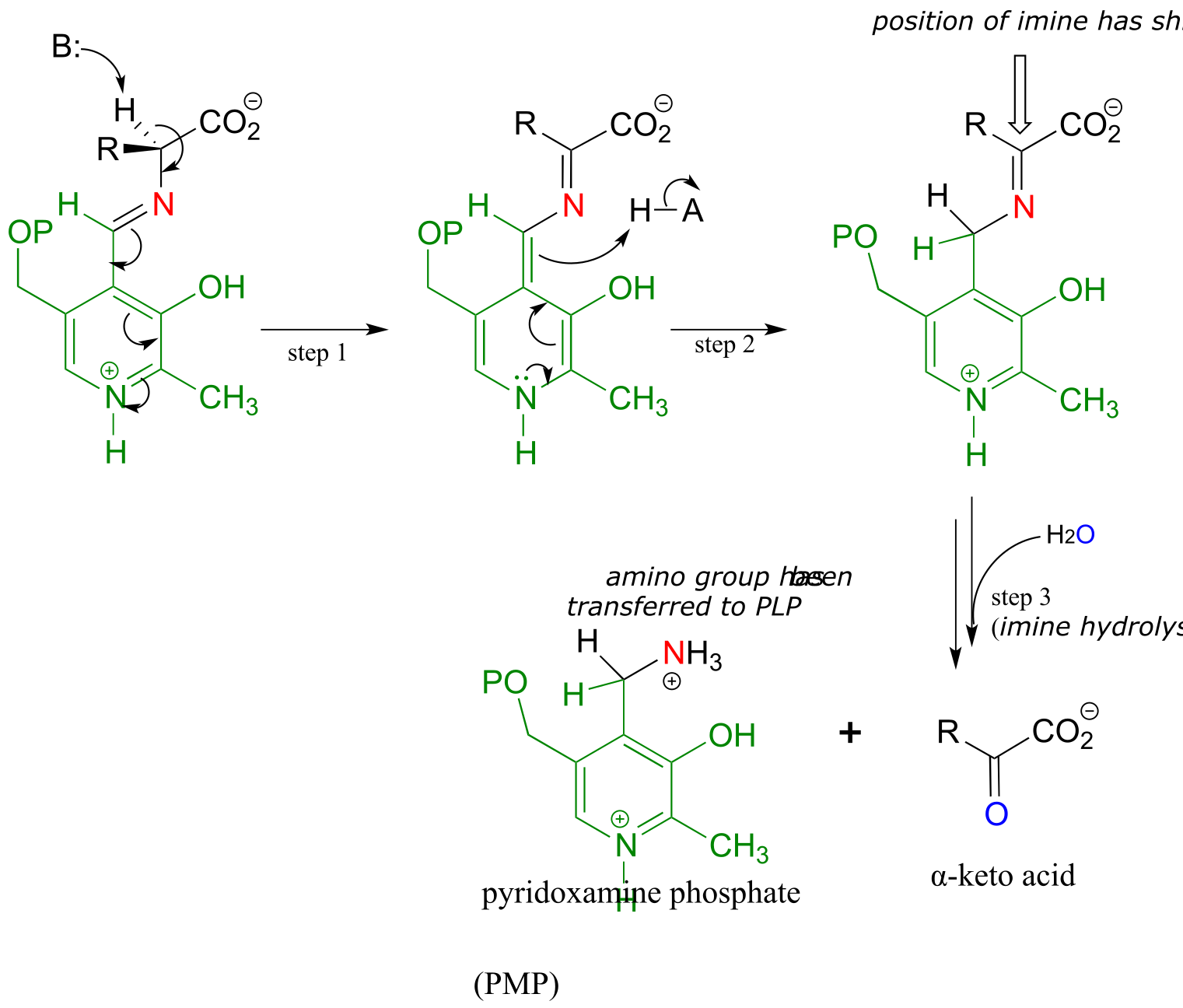
fig 16 fig 17 fig17a
Once again, step 1 is abstraction of the α-proton from the PLP-substrate adduct. However, in a transaminase reaction this initial deprotonation step is immediately followed by reprotonation at what was originally the aldehyde carbon of PLP (step 2 above), which results in a new carbon-nitrogen double bond (in other words, an imine) between the α-carbon and the nitrogen atom of the original amino acid. The repositioned imine group is then hydrolyzed (step 3 above), breaking the carbon-nitrogen bond, transferring the amino group to the coenzyme, and releasing an α-keto acid.
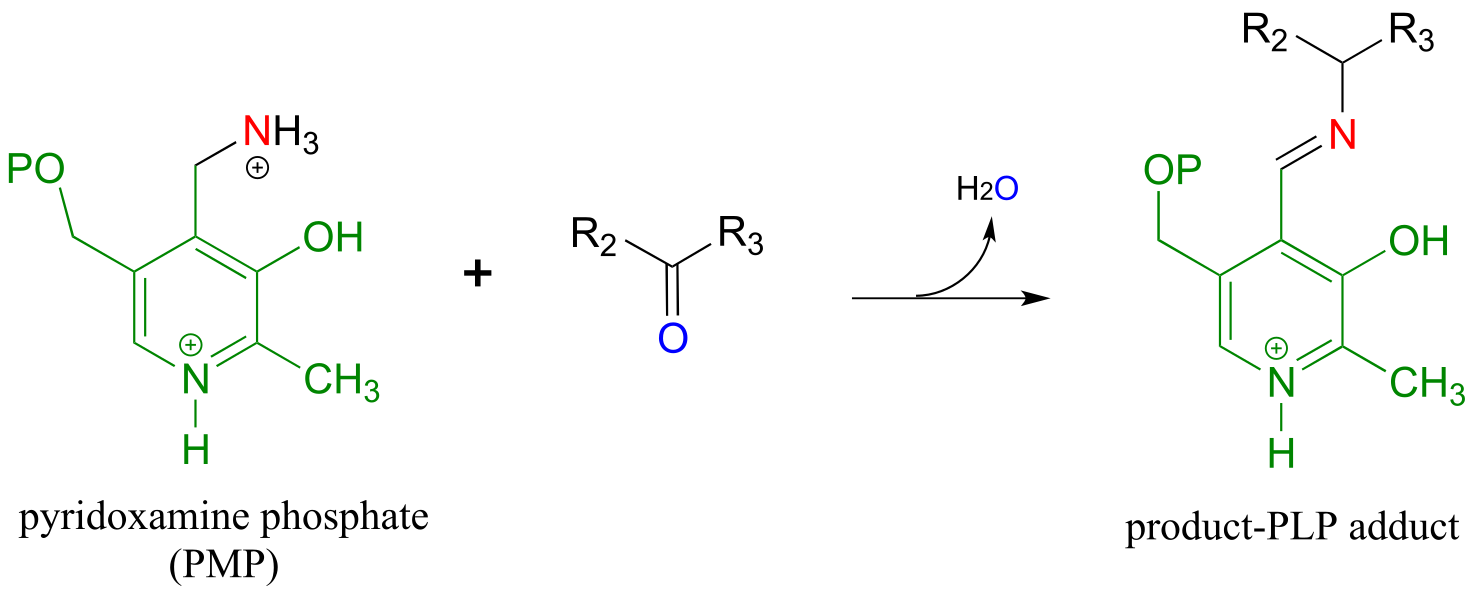
The coenzyme, which now carries an amine group and is called pyridoxamine phospate (PMP), next transfers the amine group to α-ketoglutarate (to form glutamate) through a reversal of the whole process depicted above.
Transamination reaction, phase 2
(transfer of amino group from coenzyme to acceptor molecule)
Mechanism:
see exercise below
fig 18
In a transamination reaction, the PLP coenzyme not only provides an electron sink, it also serves as a temporary ‘parking place’ for an amino group as it is transferred from donor to acceptor.
Exercise 17.2: Show a complete, step-by-step mechanism for ‘phase 2’ of the transamination reaction above.
Here is an example of a transamination reaction in the arginine biosynthesis pathway: EC 2.6.1.11

fig 19
Exercise 17.3:
a) Draw arrows for the first mechanistic step of ‘phase 2’ of the above transaminase reaction.
b) Which carbon on the substrate side of the reaction will eventually become the α-carbon of arginine?
Exercise 17.4: Propose a pathway, with three enzymatic steps, for the biosynthesis of serine from 3-phosphoglycerate. Include a generalized (‘-ase’) enzyme name for each step. Glutamate plays a role in the process as an amino group donor.
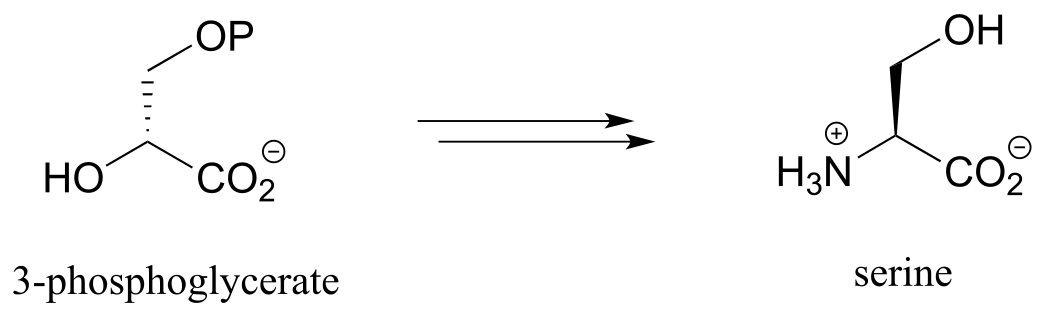
fig 20
17.1F: PLP-dependent β-elimination and β-substitution#
(Before starting this section, it would be a good idea to review E1cb β-elimination and conjugate addition reaction mechanisms in chapter 13.4)
By now it should be pretty apparent that PLP is a pretty versatile coenzyme! Two more reaction types in the PLP toolbox are β-elimination and β-substitution on amino acid substrates.
In a PLP-dependent β-elimination reaction, the coenzyme simply helps to stabilize the carbanion intermediate of the E1cb mechanism:
A PLP-dependent β-elimination reaction

Mechanism:
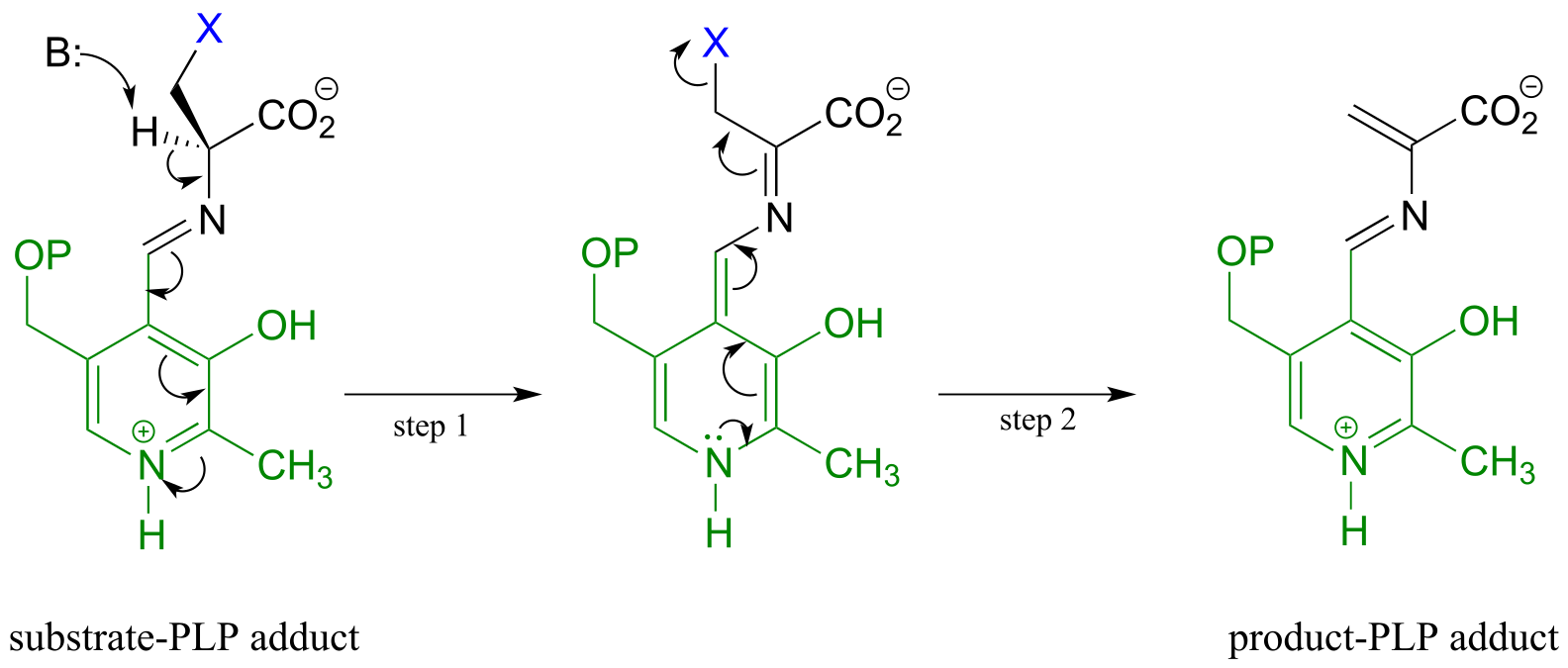
fig 21 fig 22
Serine dehydratase (EC 4.2.1.13) catalyzes a PLP-dependent β-elimination in the first step of the serine degradation pathway:
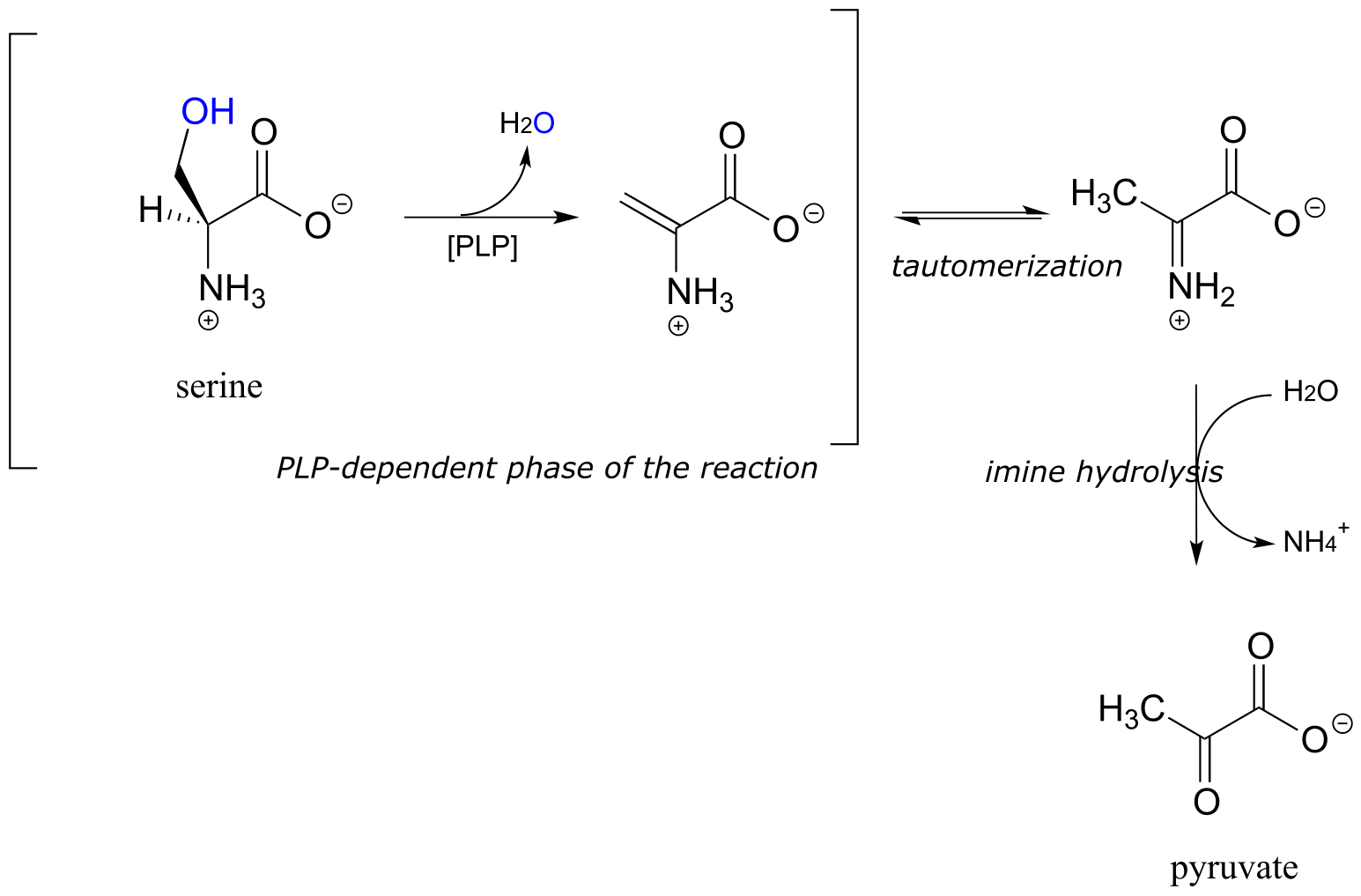
fig 23
A β-substitution reaction is simply E1cb elimination followed directly by the reverse reaction (conjugate addition) with a different nucleophile (Y in the figure below):
A β-substitution reaction:
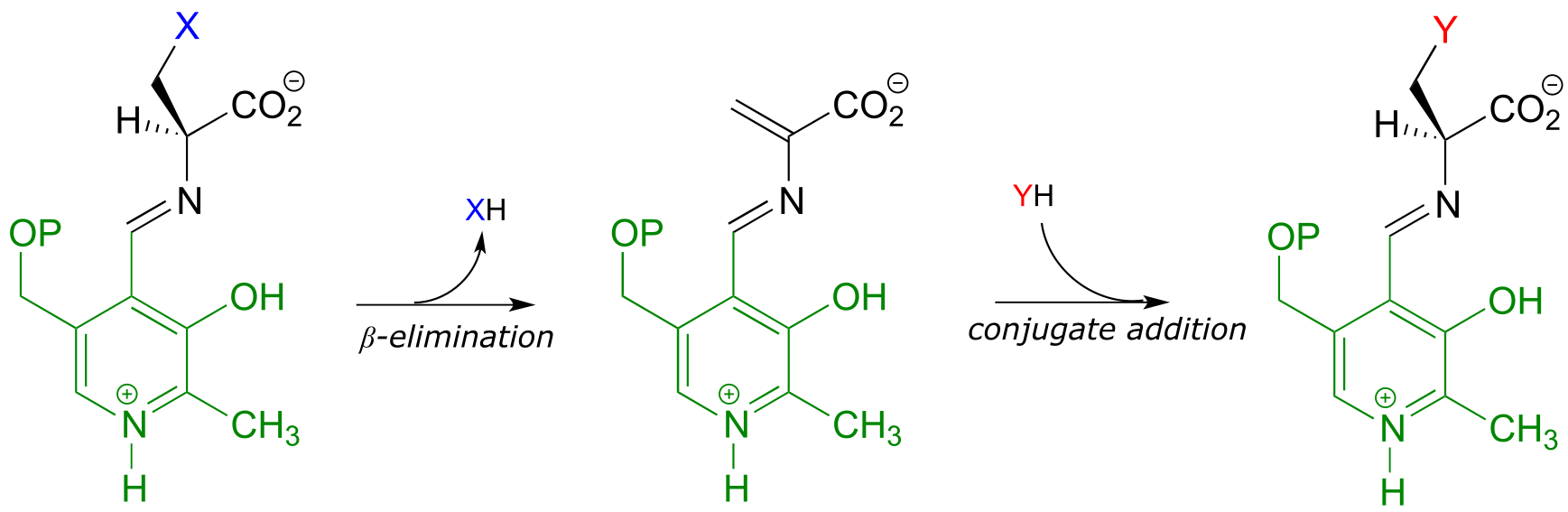
fig 24
In many bacteria, the synthesis of cysteine from serine includes a PLP-dependent β-substitution step (EC 2.5.1.47).
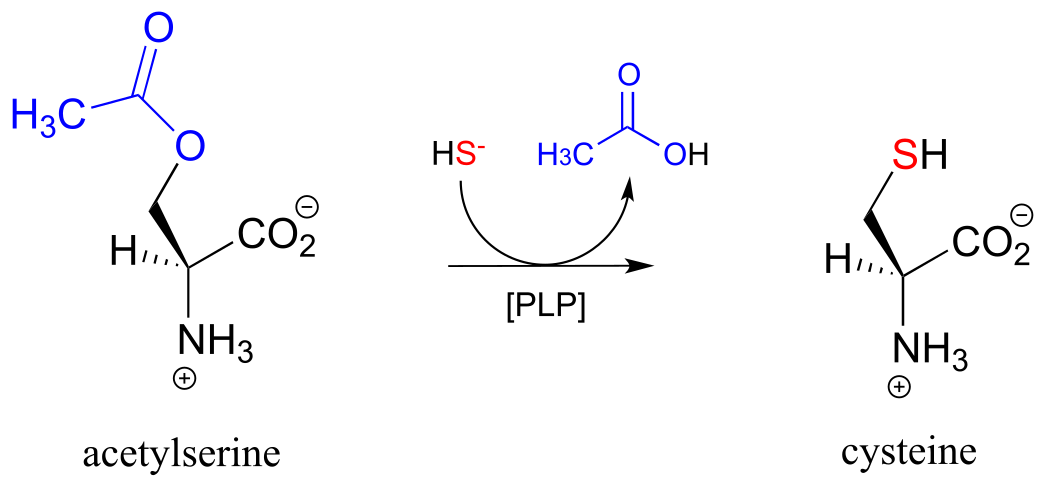
fig 25
Exercise 17.5: Draw a mechanism for the conjugate addition phase of the reaction above (end with the cysteine-PLP adduct).
17.1G: PLP-dependent γ-elimination and γ-substitution reactions#
The electron sink capability of PLP allows some enzymes to catalyze eliminations at the γ-carbon of some amino acid side chains, rather than at the β-carbon. The secret to understanding the mechanism of a γ-elimination is that PLP essentially acts as an electron sink twice - it absorbs the excess electron density from not one but two proton abstractions.
PLP-dependent γ-elimination

Mechanism:
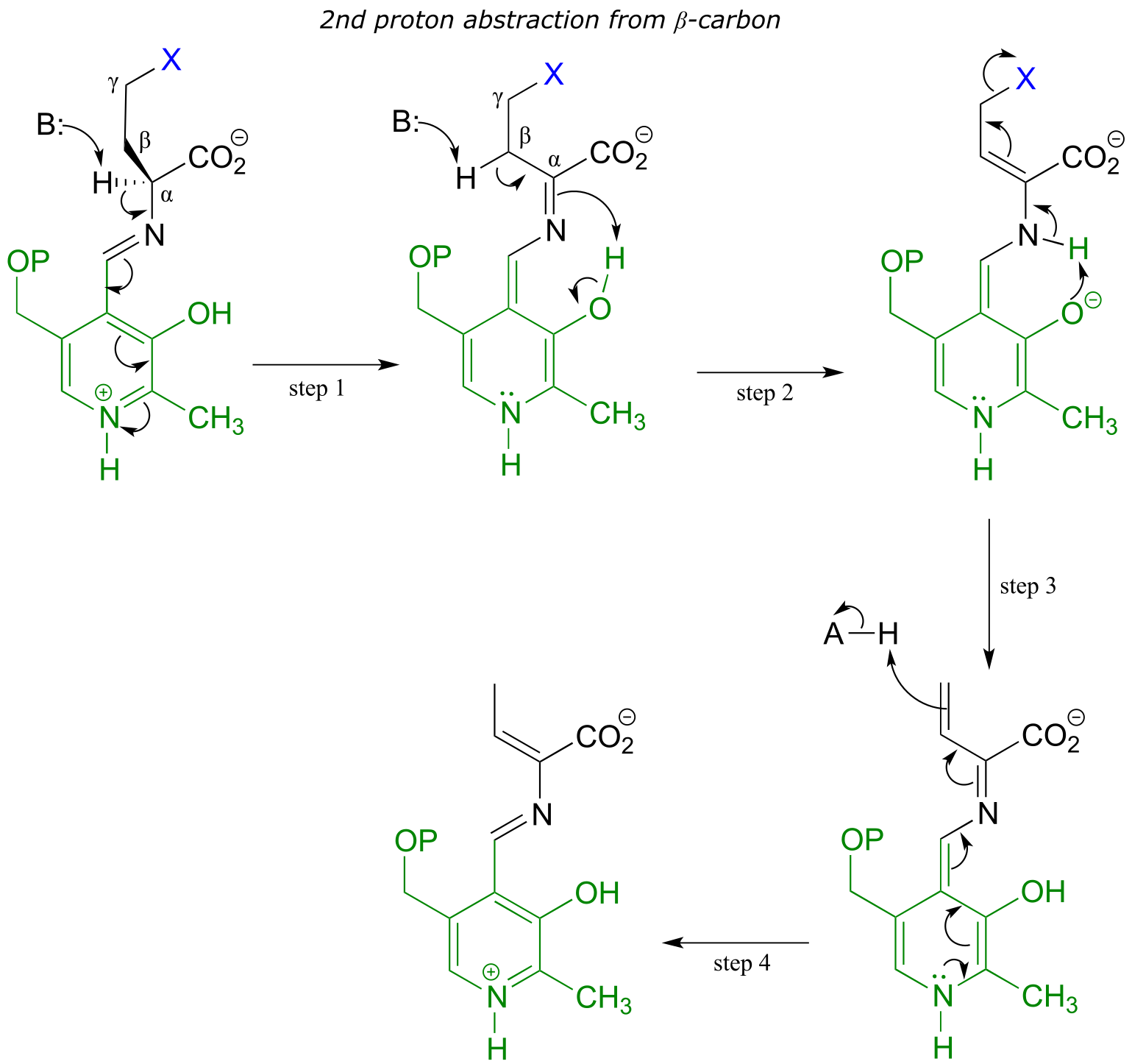
fig 26 fig 27
In a familiar first step, the α-proton of the amino acid is abstracted by an enzymatic base, and the electron density is absorbed by PLP. Next comes the new part - before anything happens to the electron density from the first proton abstraction, a second proton, this time from the β-carbon on the side chain, is abstracted, forming an enamine intermediate (step 2). The phenolic proton on the pyridoxal ring of PLP donates a proton to the nitrogen. In step 3, the leaving is expelled and a new π-bond forms between the β and γ carbons (step 3). This π-bond is short-lived, however, as the electron density from the first proton abstraction, which has been ‘stored’ in PLP all this time, flows back up to protonate the α-carbon (step 4), leaving the γ-elimination product linked to PLP via the usual imine connection.
An example is the cystathionine γ-lyase reaction in the methionine degradation pathway (EC 4.4.1.1):
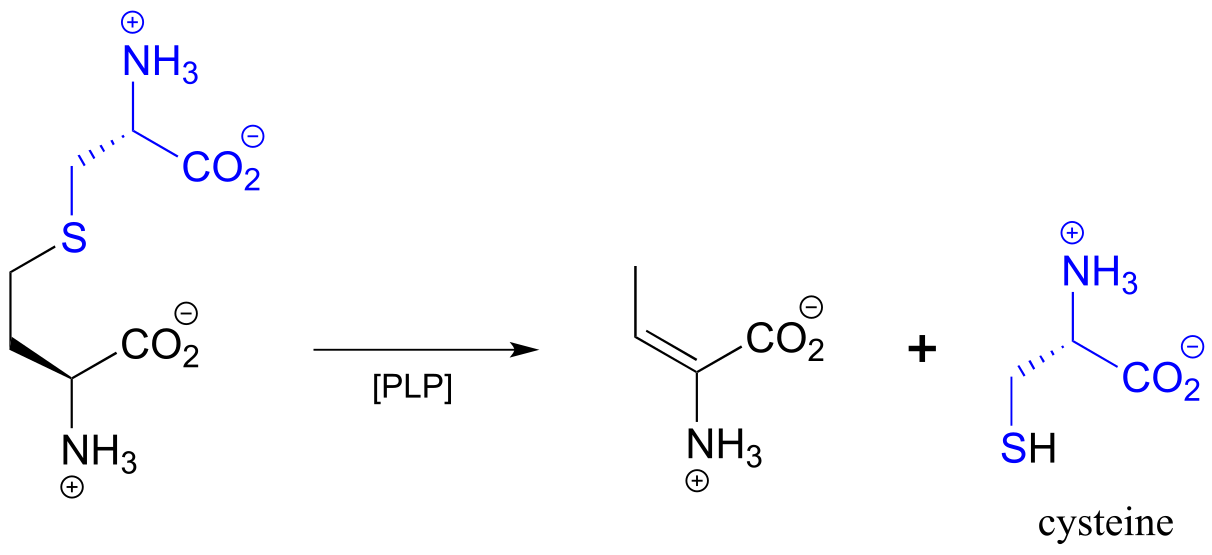
fig 28
A related reaction is PLP-dependent γ-substitution, which again is simply γ-elimination of a leaving group (X in the figure below) followed directly by the reverse process (a γ-addition) with a different nucleophile (‘Nu’ in the figure below).
PLP-dependent γ-substitution:
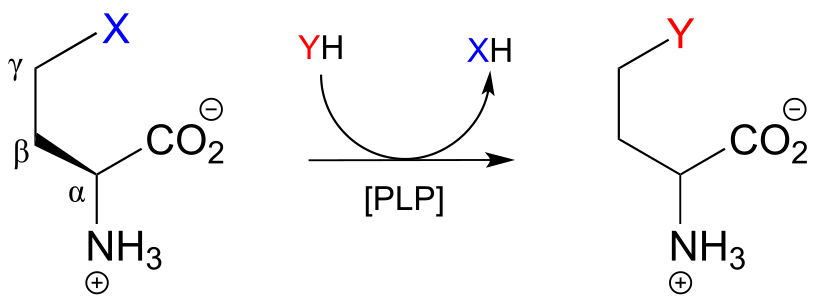
Mechanism:
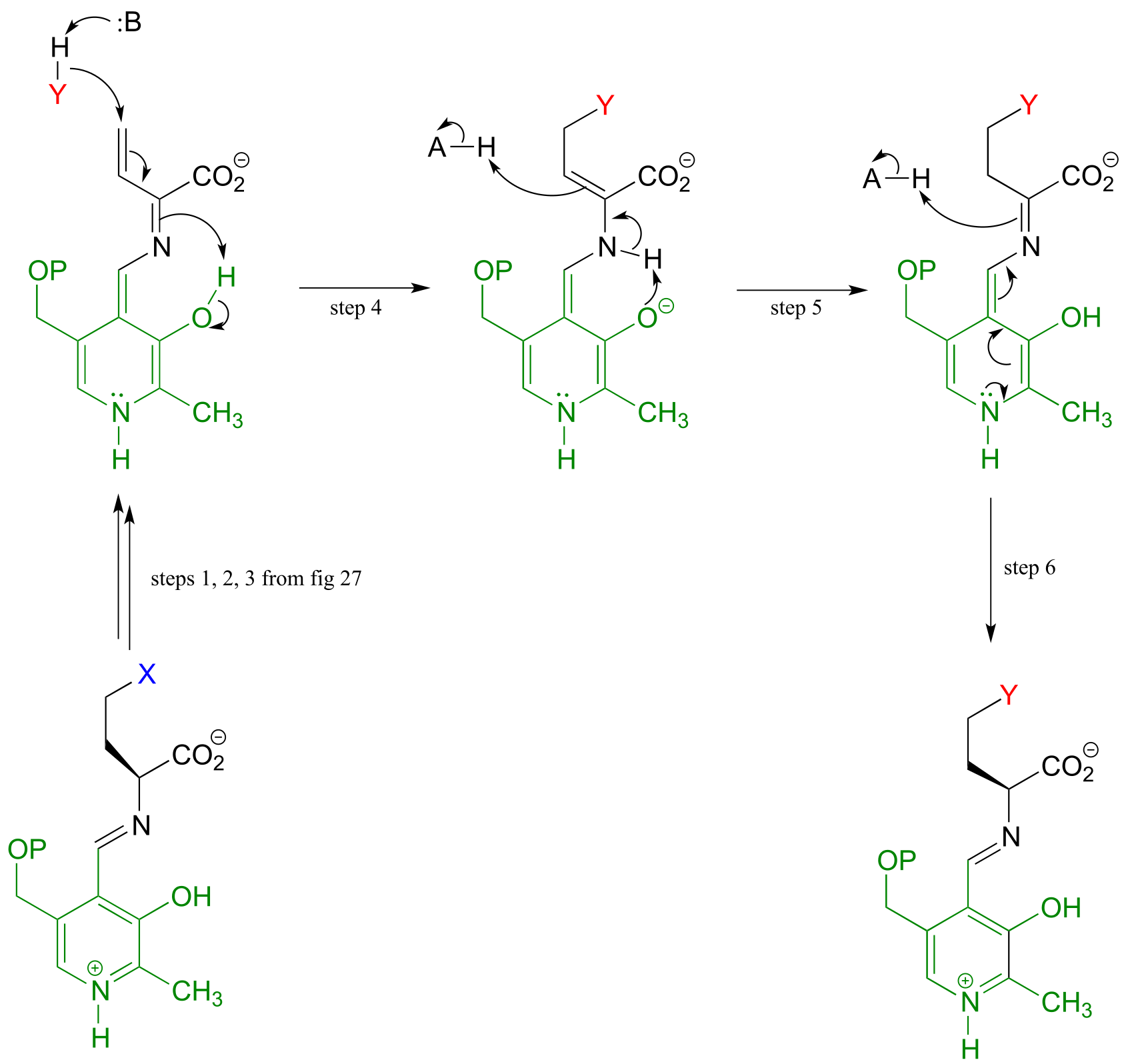
fig 29 fig 30
Below is a PLP-dependent γ-substitution reaction in the methionine degradation pathway (EC 4.2.1.22):
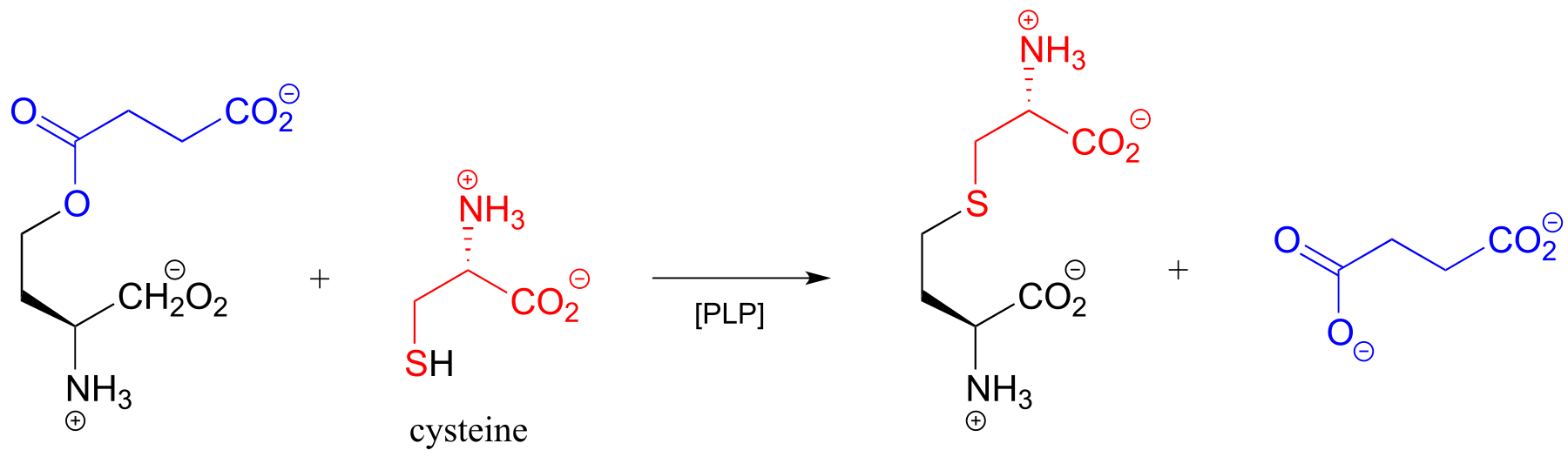
fig 31
17.1H: Racemase to aldolase: altering the course of a PLP reaction#
We have seen how PLP-dependent enzymes catalyze a variety of reaction types - racemization, retroaldol/retro-Claisen cleavage, transaminination, elimination, and substitution - which, despite their apparent diversity, are all characterized by formation of a critical carbanion intermediate which is stabilized by the ‘electron sink’ property of the PLP coenzyme. Given this common mechanistic feature, it would be reasonable to propose that the active site architecture of these enzymes might also be quite close. This idea was nicely illustrated by an experiment in which researchers found that changing a single active site amino acid of PLP-dependent alanine racemase was sufficient to turn it into a retro-aldolase (J. Am. Chem. Soc. 2003, 125, 10158).
In the ‘wild-type’ (natural) alanine racemase reaction, an active site histidine (red in figure below) deprotonates a neighboring tyrosine residue (blue), which in turn acts as the catalytic base abstracting the α-proton of the substrate. When researchers changed the tyrosine to an alanine (using a technique called ‘site-directed mutagenesis’), and substituted β-hydroxytyrosine for the alanine substrate, the new ‘mutant’ enzyme catalyzed a retro-aldol reaction.
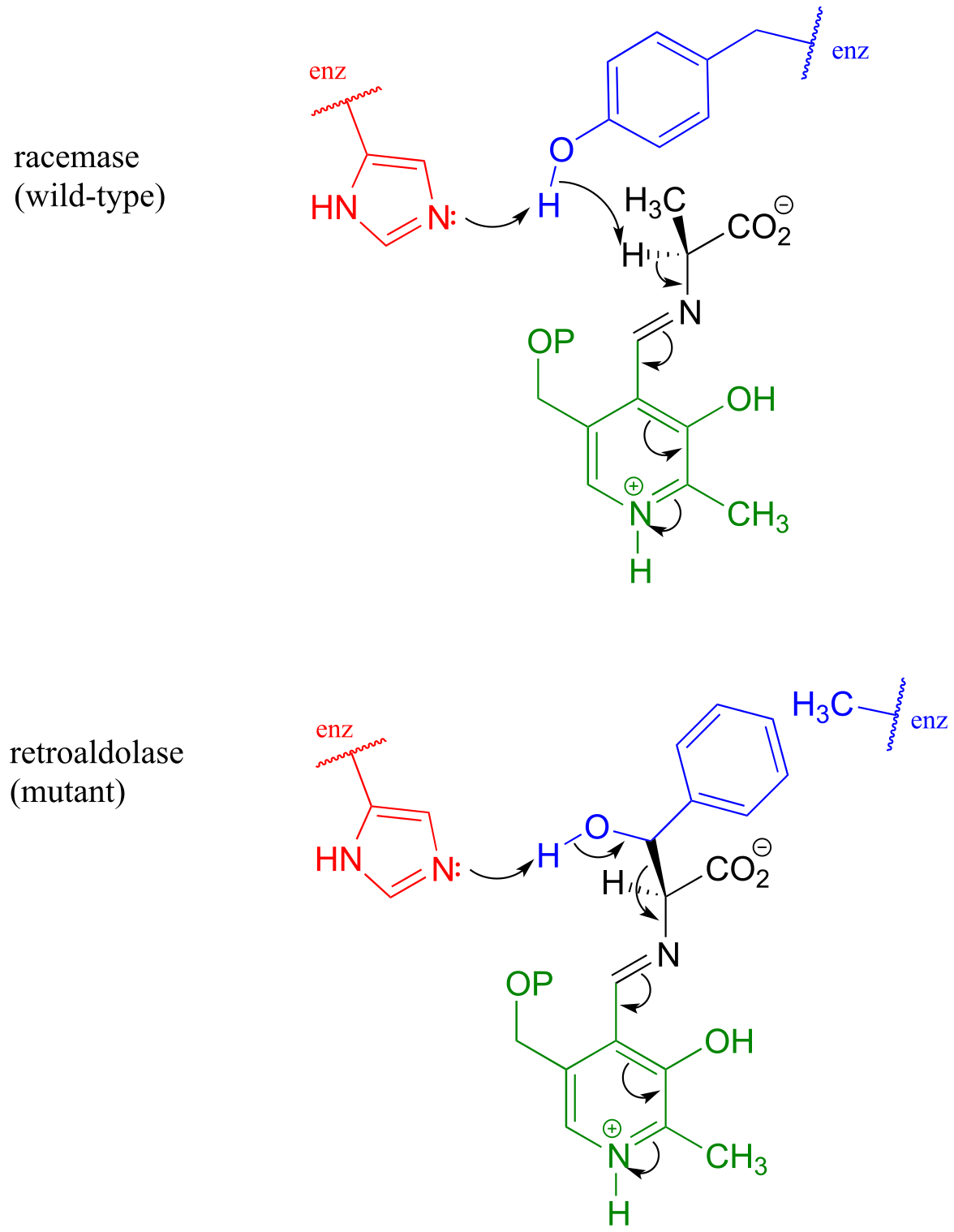
fig 32
Notice what has happened here: the basic histidine, with no tyrosine to deprotonate because of the mutation, is instead positioned to abstract a proton from the β-hydroxyl group of the new substrate, setting up a retroaldol cleavage. That was all it took to change a racemase into a retroaldolase, because the necessary PLP electron sink system was all left in place. The researchers predicted correctly that the phenyl ring of β-hydroxy tyrosine would fit nicely in the space left empty due to the tyrosine to alanine change in the mutant enzyme’s structure
These results underline the close mechanistic relationship between two PLP-dependent reactions which, at first glance, appear to be quite different - and suggest that PLP-dependent racemases and aldolases may have evolved from a common ‘ancestor’ enzyme.
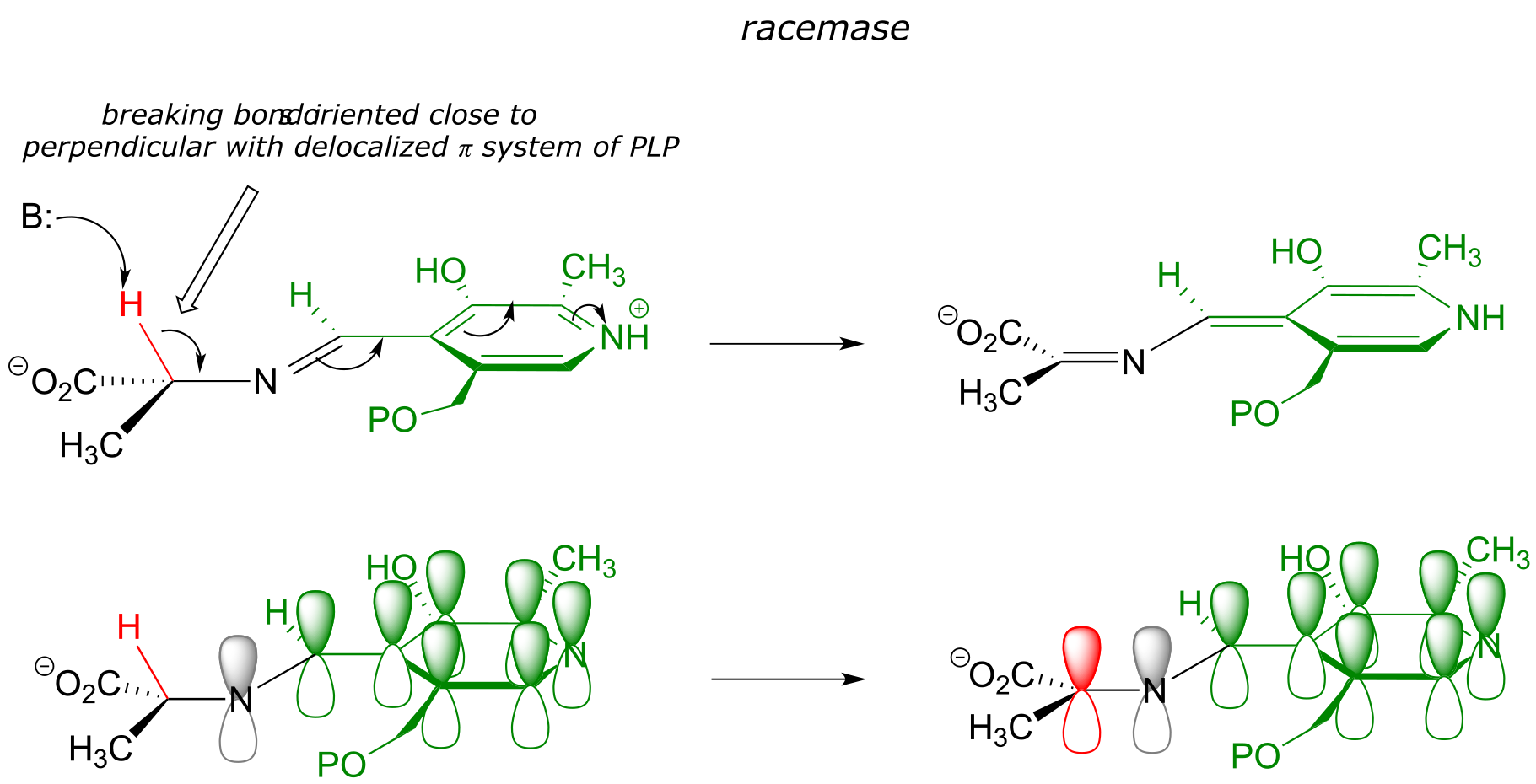
17.1I: Stereoelectronic considerations of PLP-dependent reactions#
Recall that all PLP-dependent reactions involve the cleavage of one of the bonds coming from the α-carbon of an amino acid substrate, with the coenzyme serving as an ‘electron sink’ to stabilize the intermediate that results. PLP-dependent enzymes accelerate this bond-breaking step by binding the substrate-PLP adduct in a conformation such that the bond being broken is close to perpendicular to the plane formed by the conjugated π system of PLP: this way, as the α-carbon transitions from sp3 to sp2 hybridization, the unhybridized p orbital is already oriented to overlap with the rest of the conjugated system. For example, in alanine racemase the first step is cleavage of the Cα-H bond, so it must be that bond which is positioned near-perpendicular to the PLP plane:
Likewise, in an amino acid decarboxylase, the Cα-carboxylate bond is held near-perpendicular to the PLP plane, and in hydroxymethyltransferase, the Cα-Cβ bond is in the perpendicular orientation:
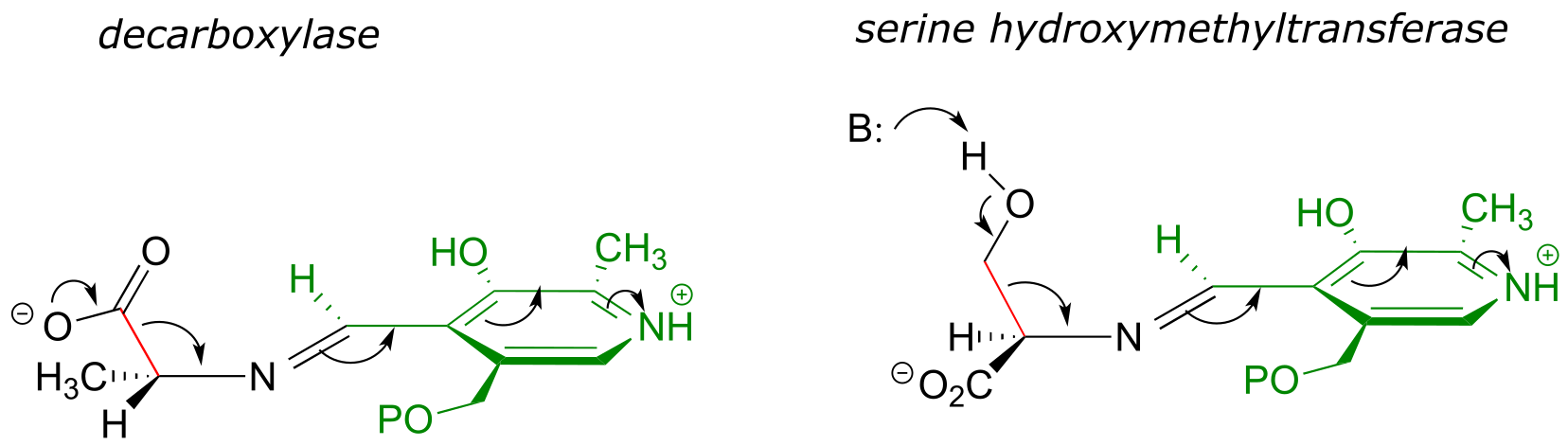
These are all good examples of how enzymatic catalysis is achieved, in part, by the ability of the active site to bind the substrate molecule in a specific conformation which contributes to the lowering of the activation energy of a key reaction step.
Section 17.2: Thiamine diphosphate (Vitamin B1)#
Thiamine diphosphate (ThDP, sometimes also abbreviated TPP or ThPP) is a coenzyme which, like PLP, acts as an electron sink to stabilize key carbanion intermediates. The most important part of the ThDP molecule from a catalytic standpoint is its thiazole ring.
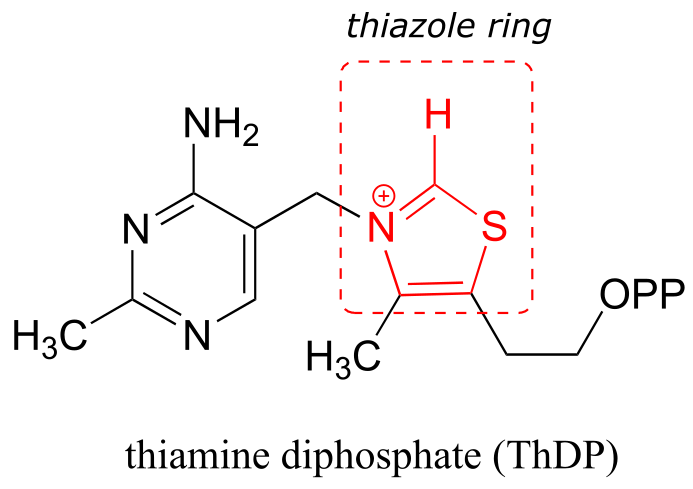
fig 33
The proton on the carbon between nitrogen and sulfur on the thiazole ring is weakly acidic, with a pKa of about 18.
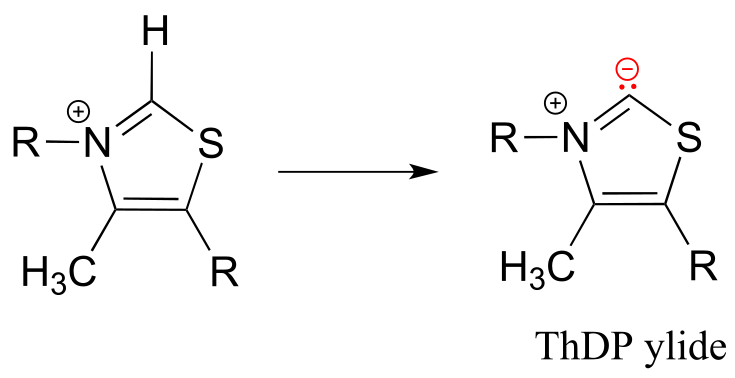
fig 34
The reason for its acidity lies partly in the ability of the neighboring sulfur atom to accept, in its open d-orbitals, some of the excess electron density of the conjugate base. Another reason is that the positive charge on the nitrogen helps to stabilize the negative charge on the conjugate base. The deprotonated thiazole is called an ylide, which is a general term for a species with adjacent positively and negatively charged atoms.
The negatively charged carbon on the ylide form of ThDP is nucleophilic, and as we shall soon see, the first step of most TPP-dependent reactions is nucleophilic attack of the ylide carbon on a carbonyl group of the reaction substrate.
ThDP plays a key role in a variety of reaction types, but the common theme in all ThDP-dependent reactions is cleavage of a bond adjacent to the carbonyl carbon of a ketone or aldehyde.
Thiamine diphosphate assists in breaking bonds next to a ketone or aldehyde:

fig 35
Consider this hypothetical decarboxylation step:
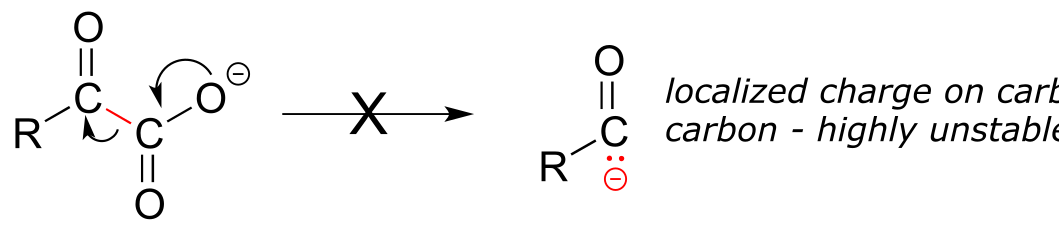
fig 37
Hopefully, you can quickly recognize that this is not a chemically reasonable step, because the intermediate species which results from decarboxylation has a negative charge localized on the ketone carbon - a very unstable, unlikely intermediate indeed.
(Recall from section 13.1 that decarboxylation steps usually result in intermediates in which the negative formal charge is delocalized to an oxygen or nitrogen - in other words, enolates or enamines.)
Now consider, however, a reaction going on in your cells right now, catalyzed by the enzyme pyruvate decarboxylase (EC 4.1.1.1):
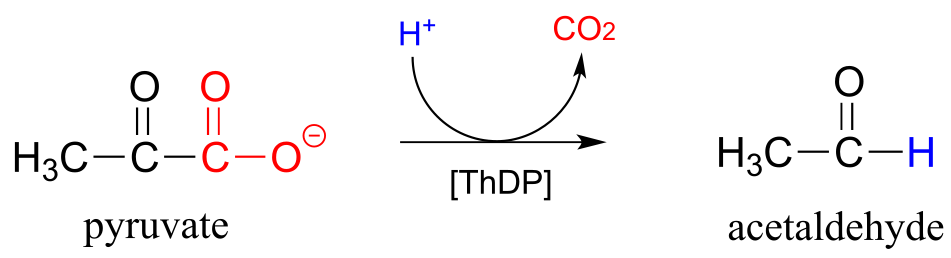
fig 36
Somehow, the enzyme manages to accomplish this ‘impossible’ decarboxylation. How does this happen? Here is where the thiamine diphosphate coenzyme comes in.
A ThDP-dependent decarboxylation reaction (pyruvate decarboxylase):
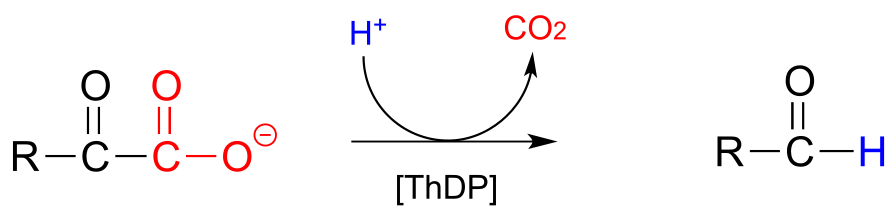
Mechanism:
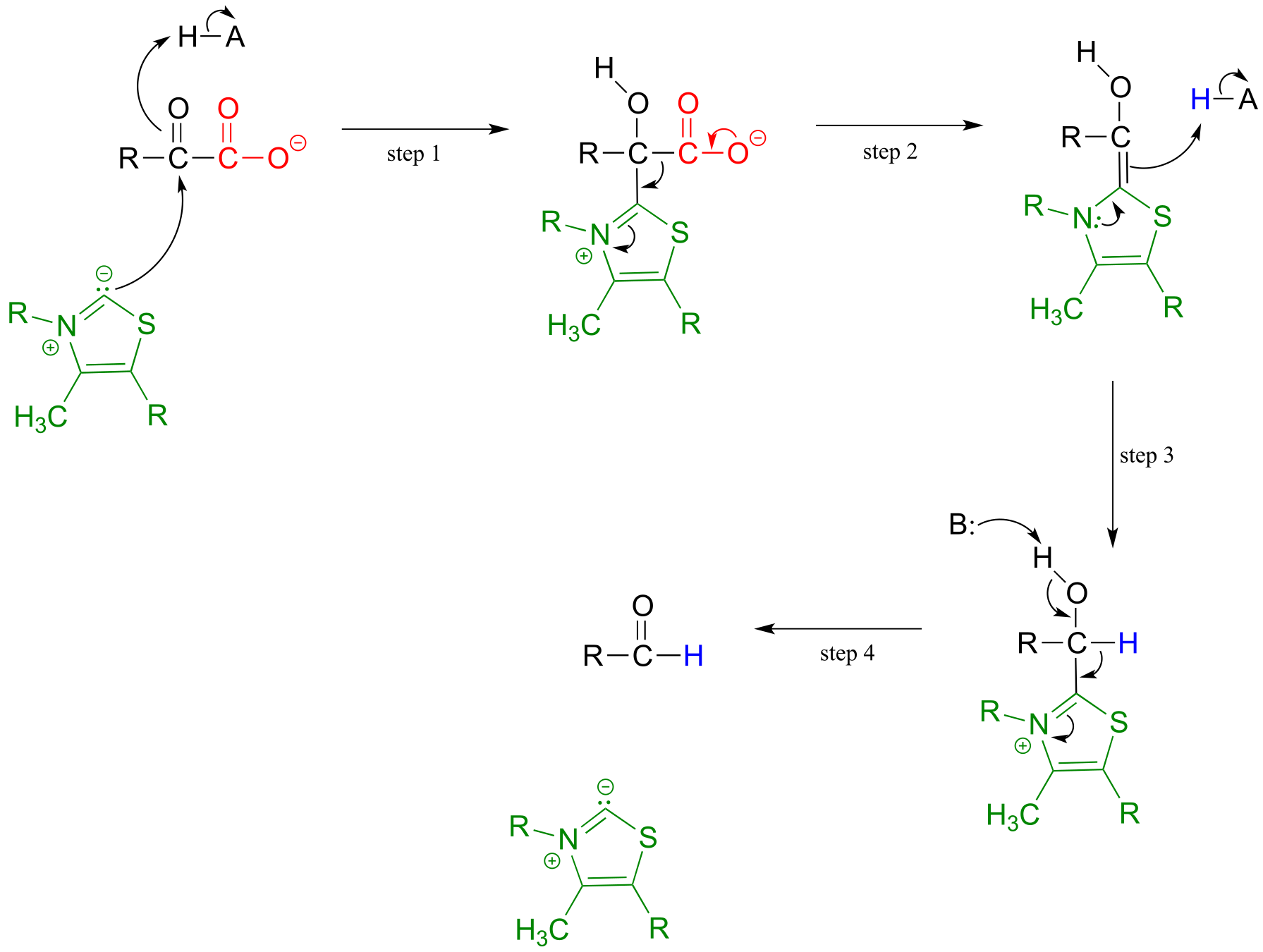
fig 36a fig 38
Upon binding to the enzyme’s active site, ThDP quickly loses a proton. The nucleophilic ylide carbon then adds to the carbonyl carbon of pyruvate.
Look carefully at the intermediate that results from step 1 in the mechanism above. The thiazole ring of ThDP, once it has added to the carbonyl of pyruvate, provides an ‘electron sink’ to absorb the electrons from decarboxylation (step 2). Note which bond is breaking in step 2 - as was mentioned earlier, the common function of ThDP is to make possible the cleavage of a bond to a carbonyl carbon.
In step 3, the electrons from decarboxylation flow back to abstract a proton from an acidic group in the active site. All that remains is for the product to break free of thiamine in step 4.
Thiamine can also assist in decarboxylation-addition reactions:
ThDP-dependent decarboxylation-addition:
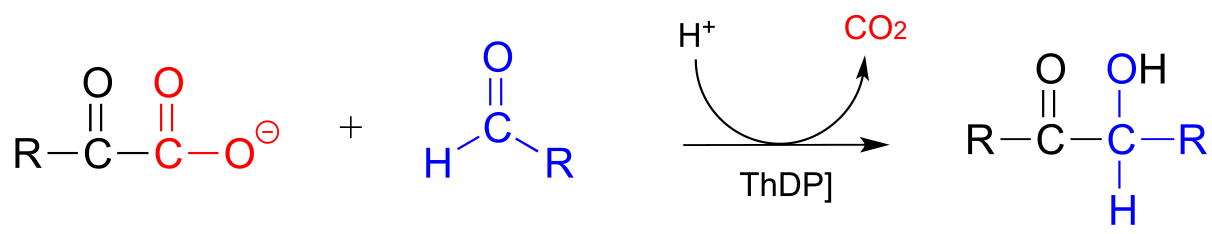
Mechanism:
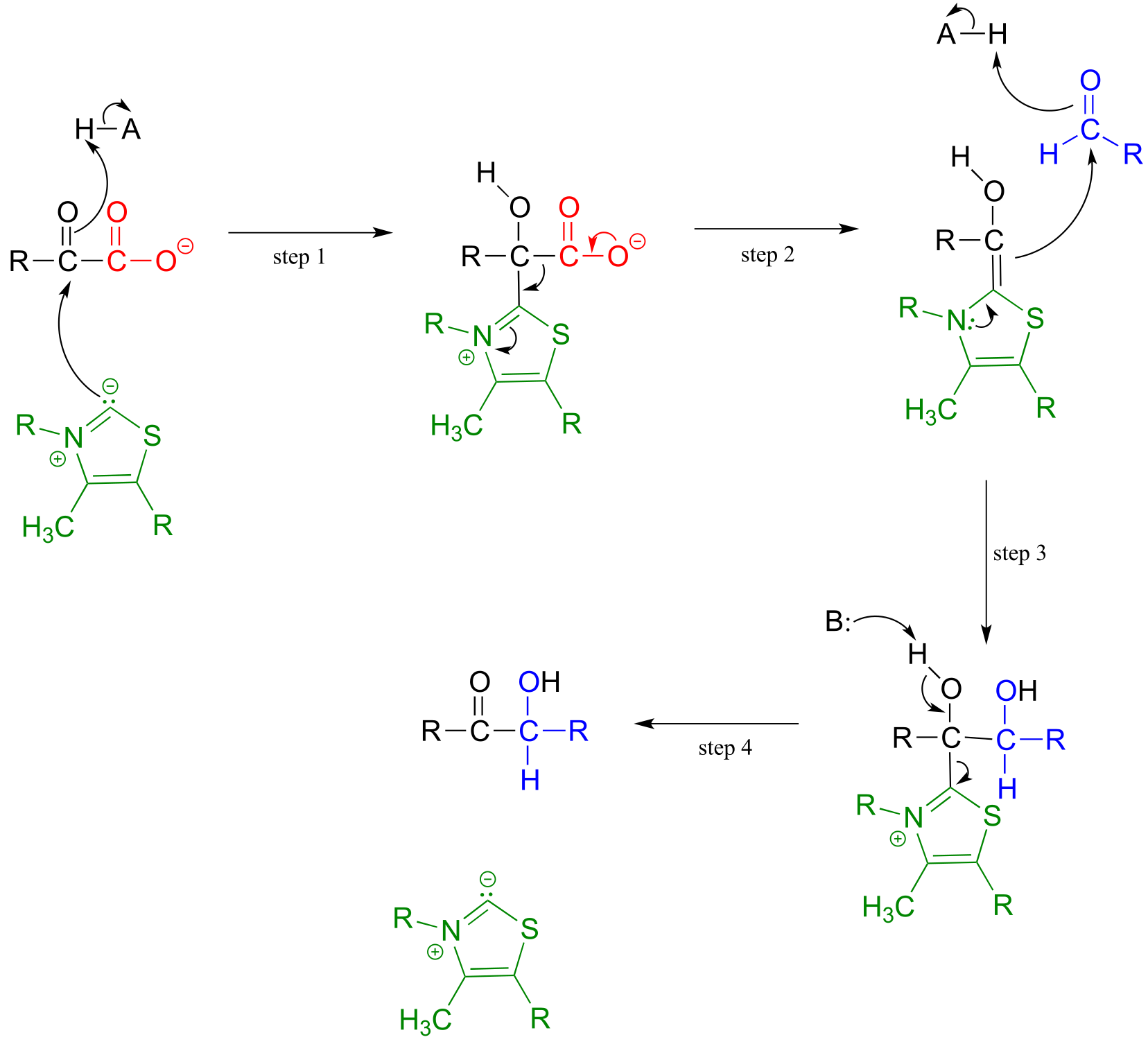
fig 39a fig 39
Here, the electron-rich intermediate formed from the decarboxylation step (step 2) simply goes on to act as a nucleophile rather than as a base, adding to the carbonyl group of an aldehyde or ketone (step 3). As before, the product breaks free of ThDP in step 4.
An example is the first step in the biosynthetic pathway for isoprenoid compounds in bacteria:
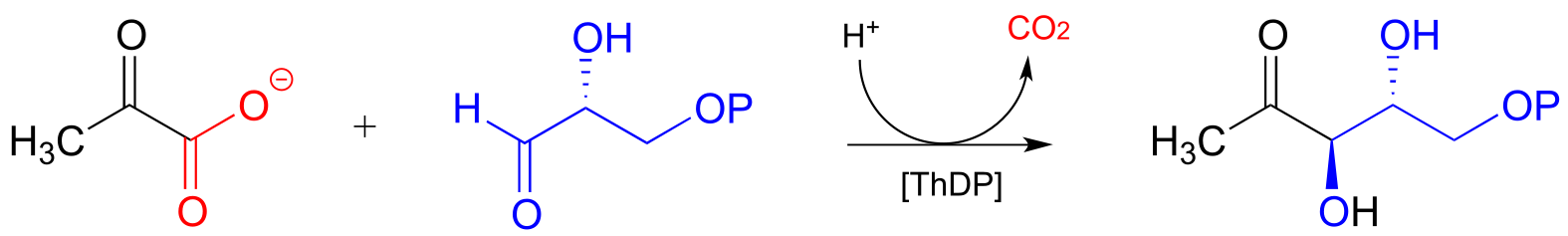
fig 40
Transketolase, a ThDP-dependent enzyme in the pentose phosphate pathway of sugar metabolism, catalyzes a carbon-carbon bond break step, followed by a carbon-carbon bond forming step. The substrates and products are at similar energy levels, so the reaction is completely reversible.
Transketolase reaction:
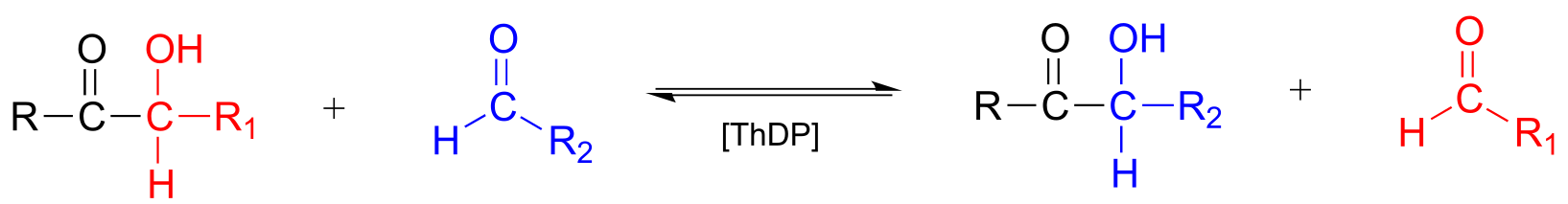
Mechanism:
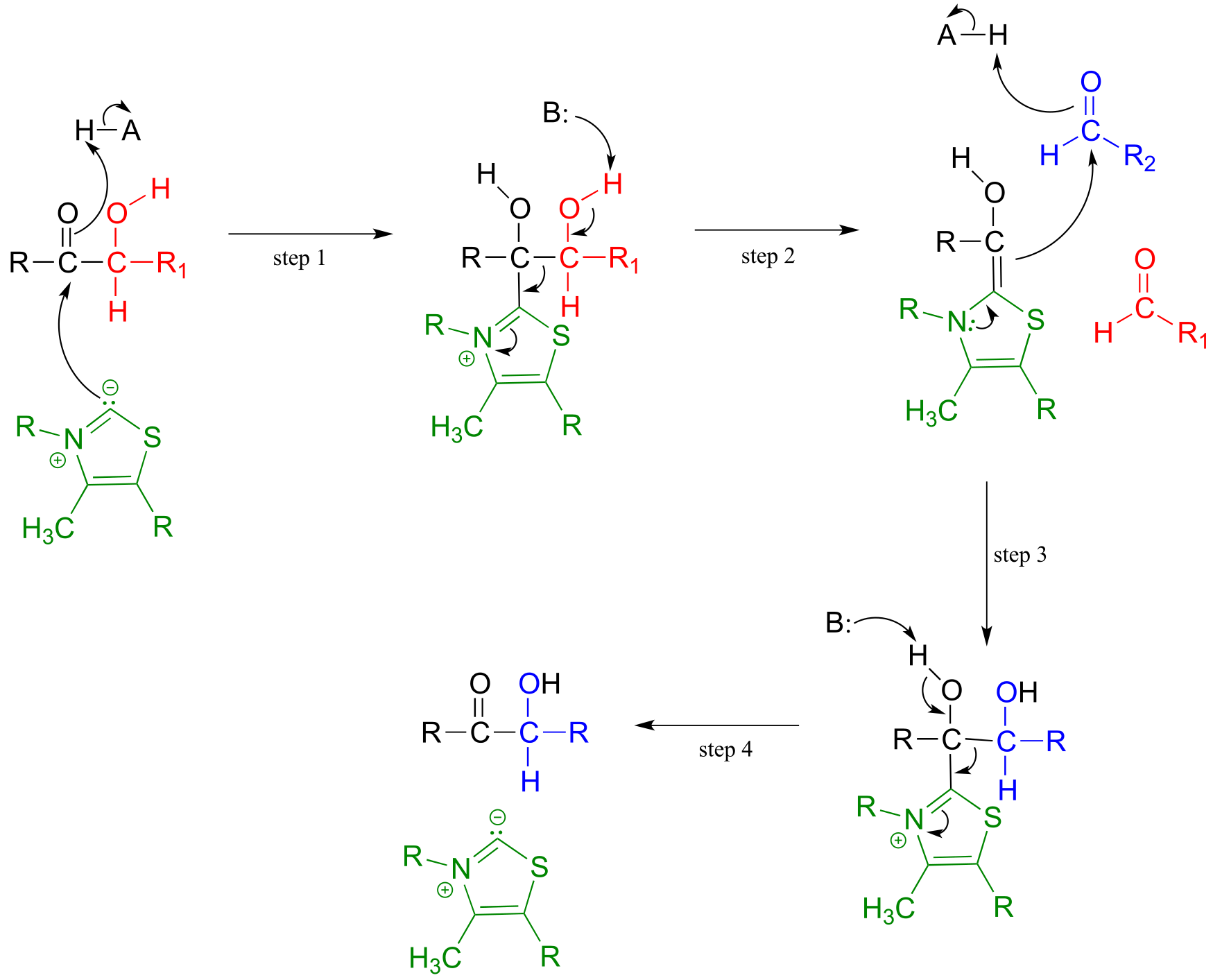
Below is an actual example of a transketolase-catalyzed transformation from the pentose phosphate pathway (shown in Fischer projections, as is common for sugar structures).
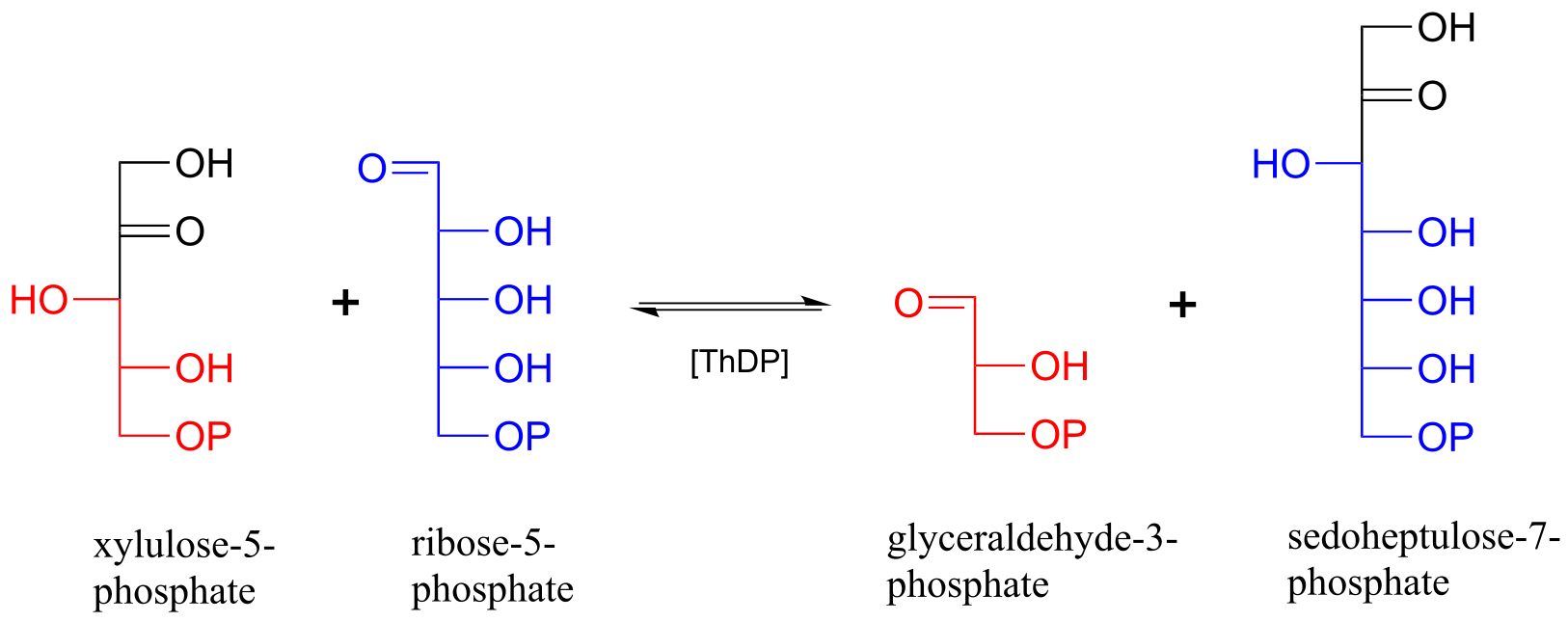
fig 43
Exercise 17.6: As was mentioned above, the transketolase reaction is highly reversible. Do you think the same can be said for the decarboxylation and decarboxylation-addition reactions we saw in this section? Why or why not?
Exercise 17.7: (Challenging!) Propose a mechanism for the reaction below. Hint: This is a ThDP-facilitated decarboxylation/ Michael addition, followed by E1cb elimination of pyruvate. A Michael addition is the name for a conjugate addition with a carbon nucleophile. (J. Mol. Biol. 2010, 401, 253).
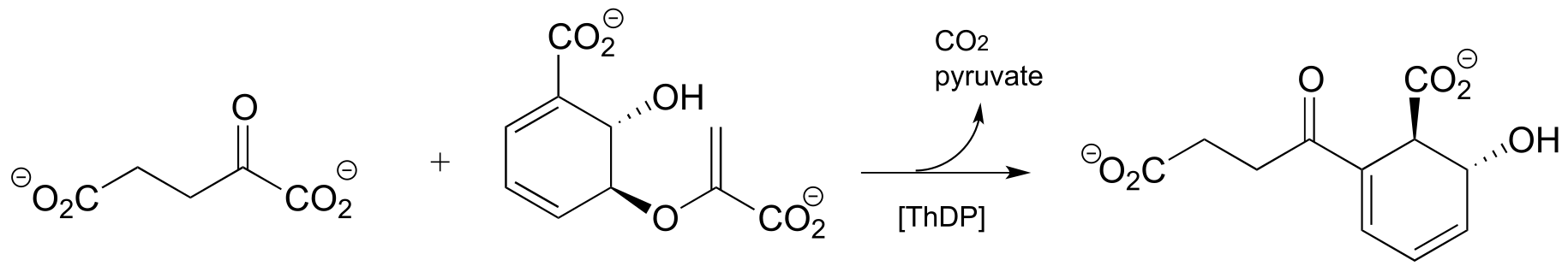
fig 44
Exercise 17.8: Propose a mechanism for the reaction below. Hint: the mechanism can be described as a ThDP-facilitated dehydration step, followed by a tautomerization step, followed by a hydrolytic expulsion of ThDP (a different kind of ThDP expulsion from what we have seen so far!)
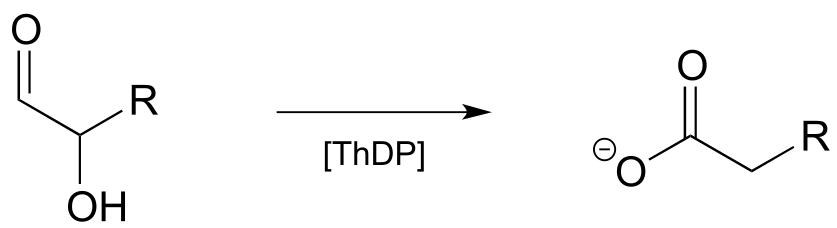
fig 44a
Section 17.3: Thiamine diphosphate, lipoamide and the pyruvate dehydrogenase reaction#
The enzyme pyruvate dehydrogenase is one of the most central of all the enzymes of central metabolism: by converting pyruvate to acetyl-CoA, it links glycolysis (where glucose is broken down into pyruvate) to the citric acid cycle, into which carbons enter in the form of acetyl-CoA. Five ceonzymes are involved: coenzyme A, nicotinamide, thiamine diphosphate, FAD, and finally lipoamide, one which is new to us at this point.
Reaction catalyzed by pyruvate dehydrogenase:
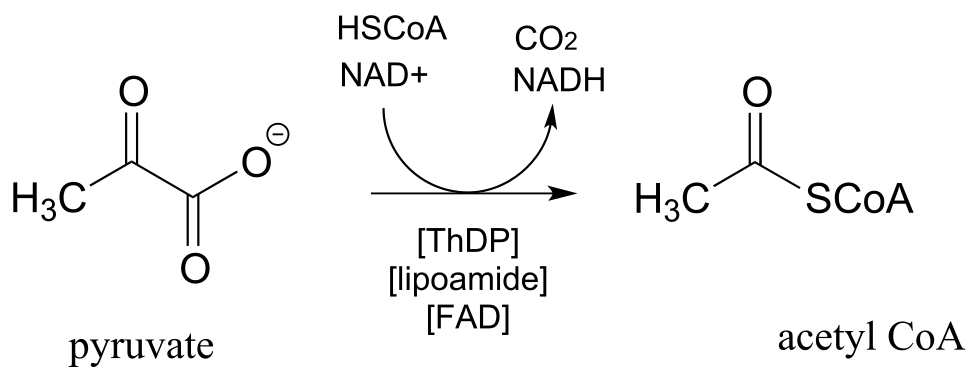
fig 45
You will learn more about the structure and metabolic role of this complex and remarkable enzyme in a biochemistry course. Here, we will focus on the multi-step organic reaction it catalyzes, which we are at long last equipped to understand.
Looking at the reaction, you should recognize that, first of all, the pyruvate substrate is being oxidized - it starts out as a ketone, and ends up as a thioester, losing carbon dioxide in the process. Ultimately, the oxidizing agent in this reaction is NAD+, but the reduction of NAD+ is linked to the oxidative decarboxylation of pyruvate by FAD and a disulfide-containing coenzyme called lipoamide, which is lipoic acid attached by an amide linkage to a lysine residue on the enzyme.
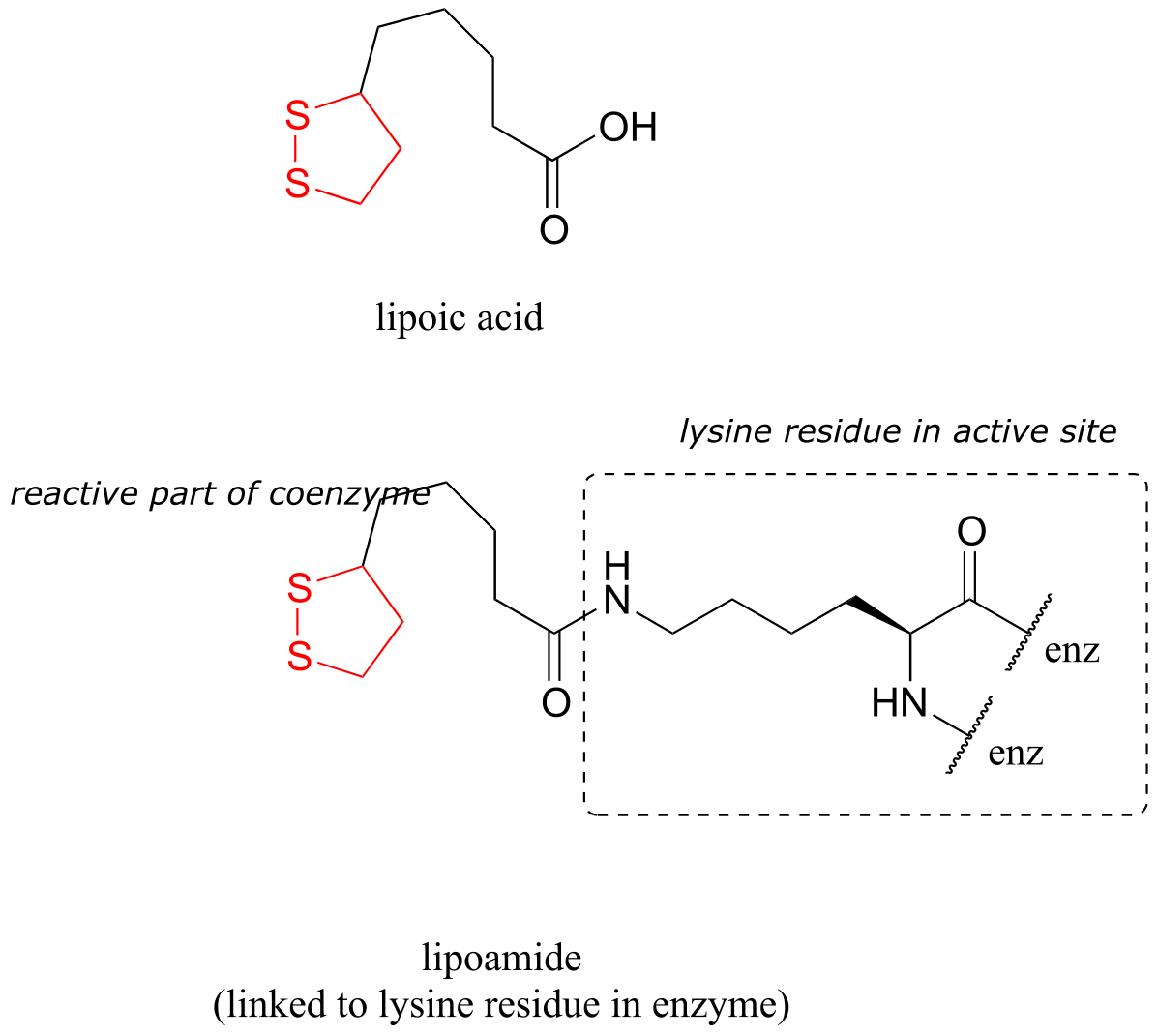
fig 45a
The second thing to notice is that, because the reaction involves breaking the bond between the ketone carbon and an adjacent carbon, thiamine diphosphate (ThDP) coenzyme is required. In fact, the first phase of the reaction (steps 1 and 2 below) is identical to that of pyruvate decarboxylase, an enzyme we discussed a few pages ago.
The pyruvate decarboxylase reaction mechanism
Phase 1: Decarboxylation of pyruvate
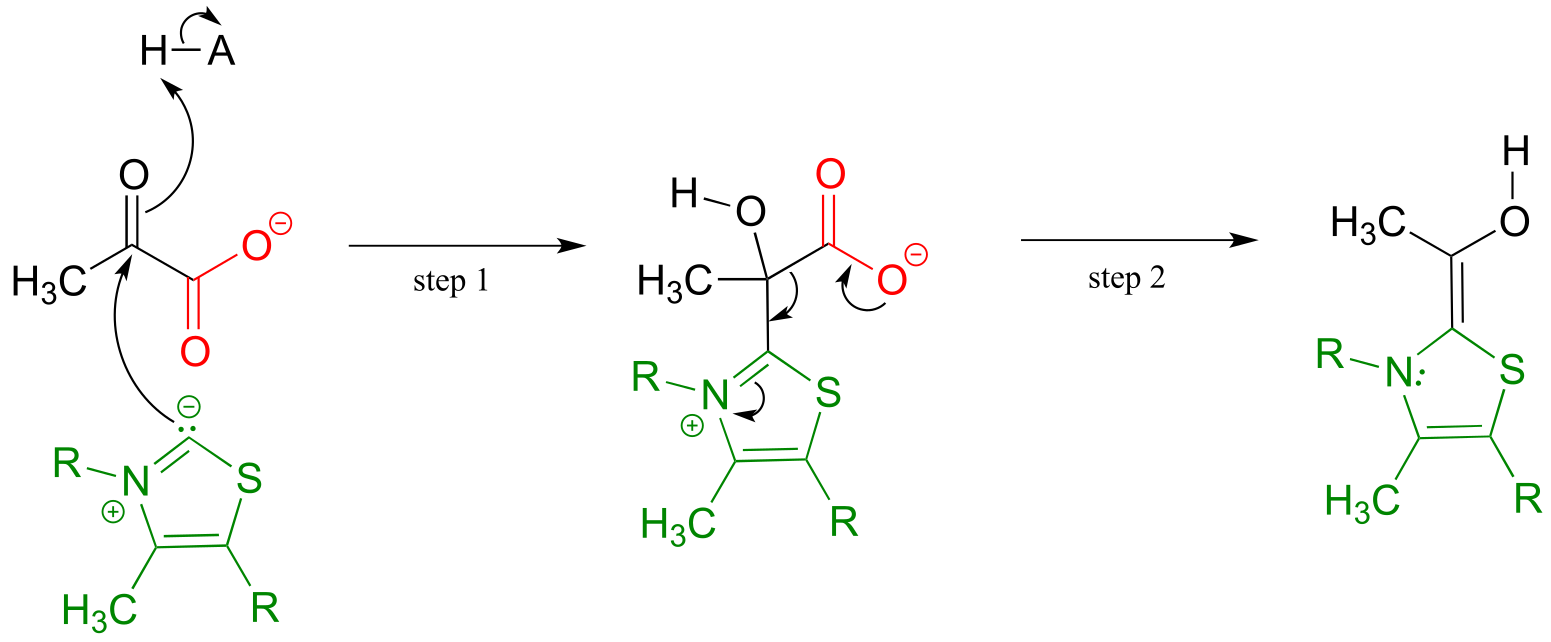
fig 46
The ThDP-stabilized carbanion then acts as a nucleophile, cleaving the disulfide bridge of lipoamide (step 3 below). It is in this step that oxidation of the substrate is actually occurring. After the resulting thioester product is released from ThDP (step 4 below), it undergoes transesterification form acetyl-CoA, the product of the reaction.
Phase 2 of the pyruvate decarboxylase reaction mechanism: lipoamide-mediate oxidation to acetyl-CoA
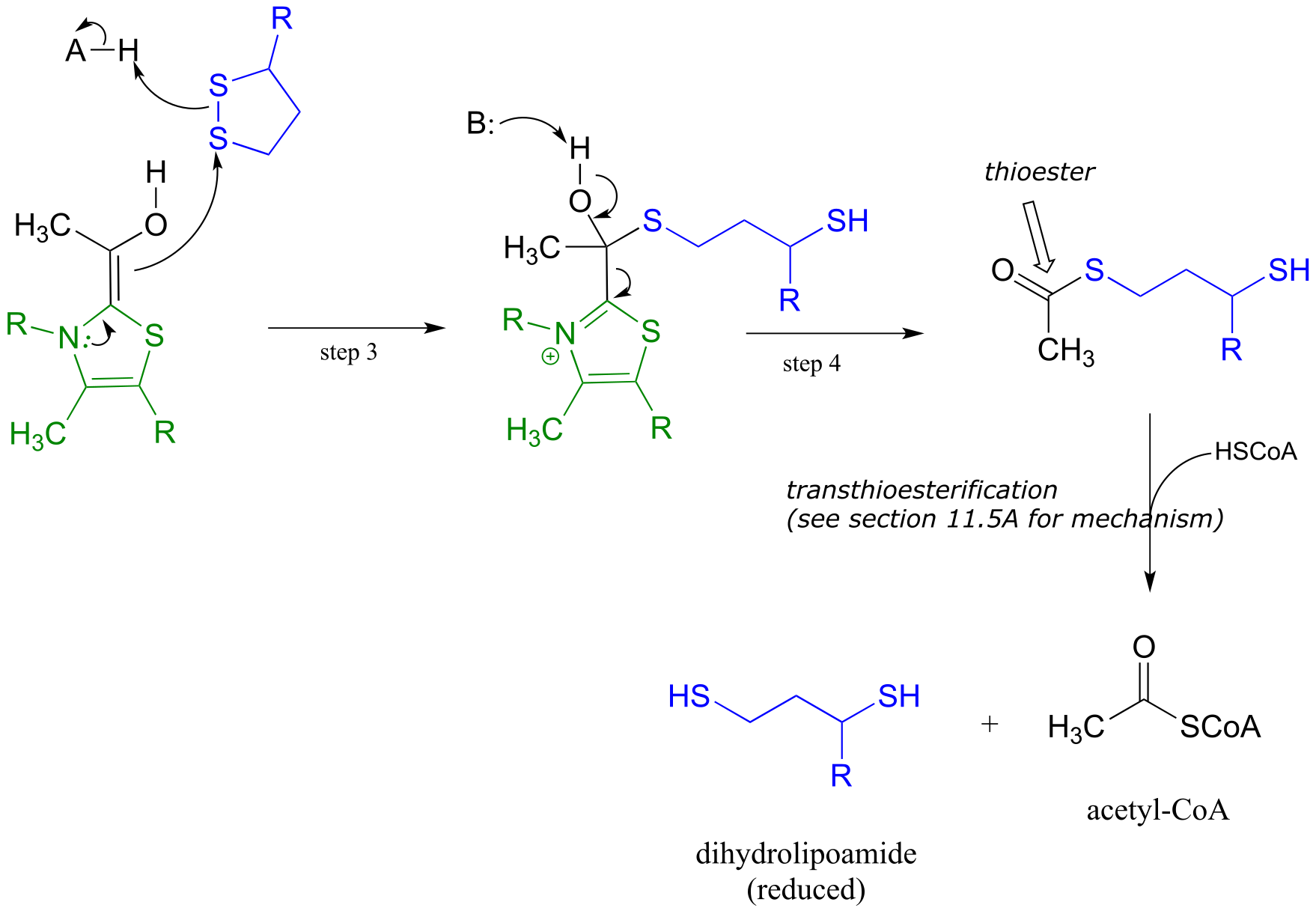
fig 47
We are not done yet! In order for the catalytic cycle to be complete, the reduced dihydrolipoamide must be regenerated back to its oxidized state through disulfide exchange with a disulfide bond on the enzyme. The pair of enzymatic cysteines is then oxidized back to disulfide form by an FAD-dependent reaction.
Phase 3 of the pyruvate decarboxylase reaction mechanism: regeneration of lipoamide
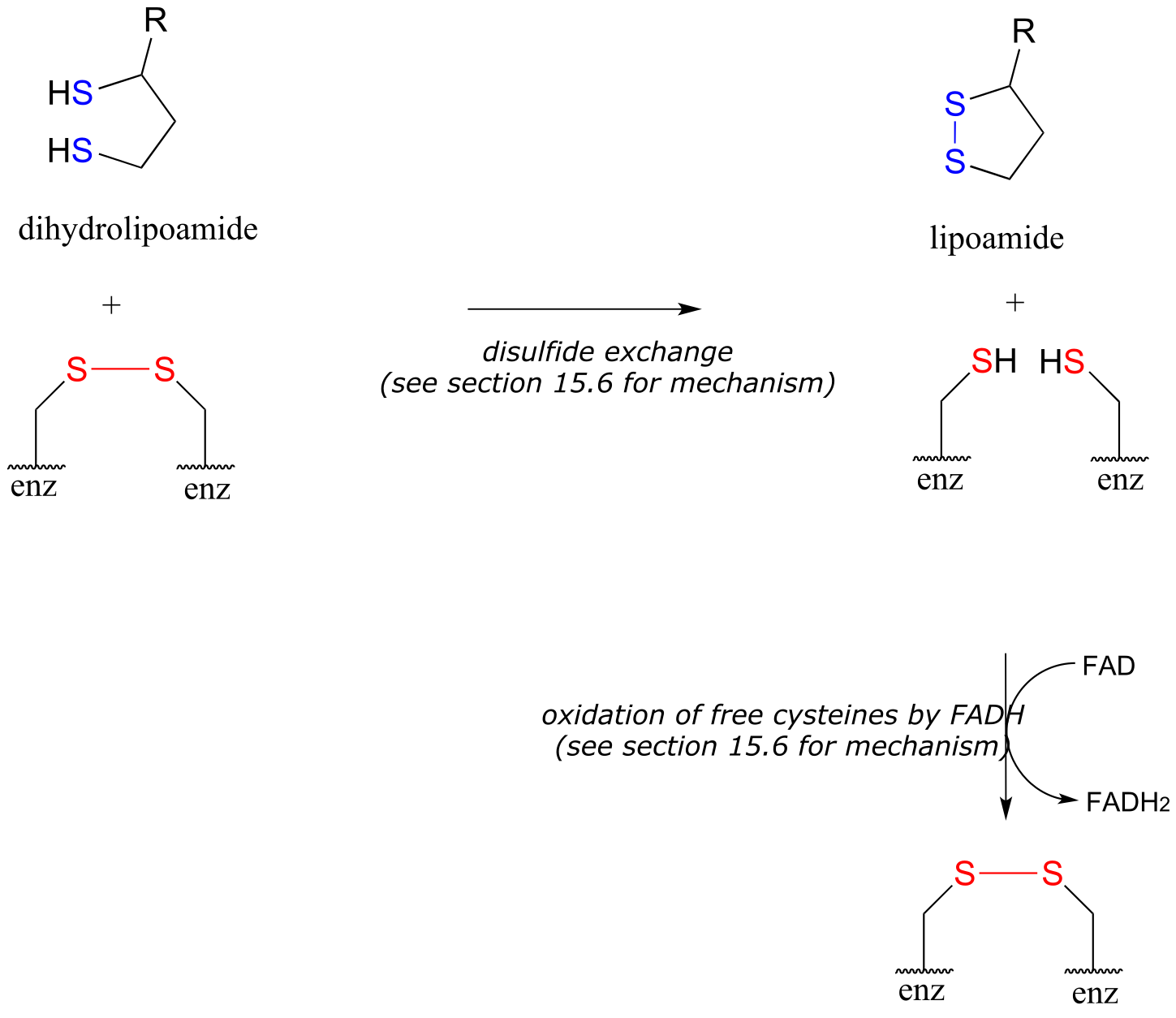
fig 48
Finally, FAD is regenerated with concurrent reduction of NAD+:
Phase 4: Regeneration of FADH2:
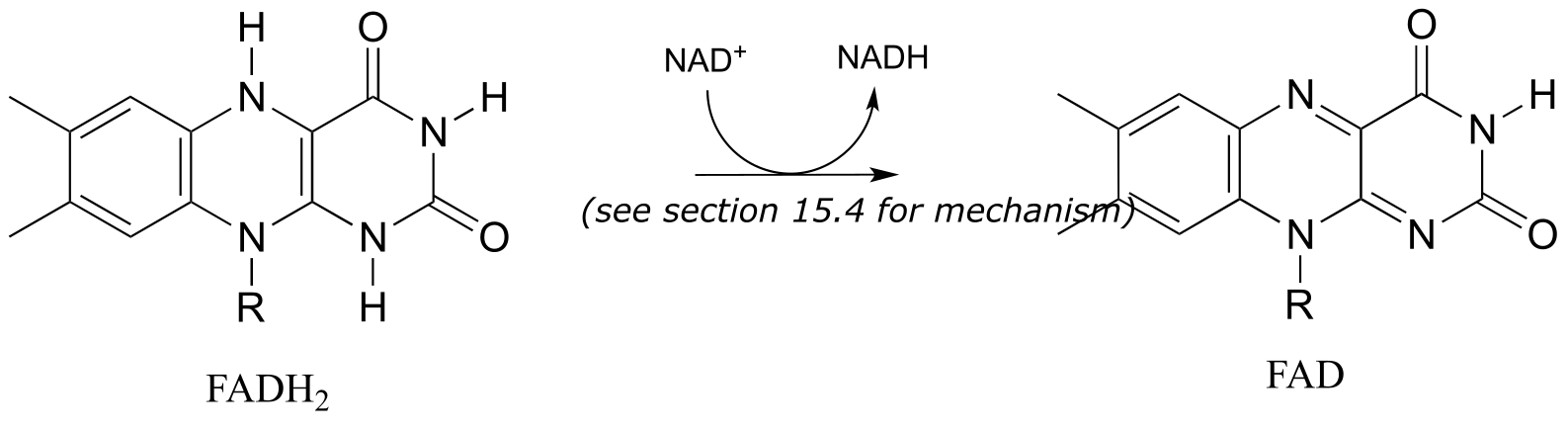
fig 49
Section 17.4: Folate#
Folate, or vitamin B9, is essential for a variety of important reactions in nucleotide and amino acid metabolism. The reactive part of folate is the pterin ring system, shown in red below. The conventional atom numbering system for folate is also indicated.
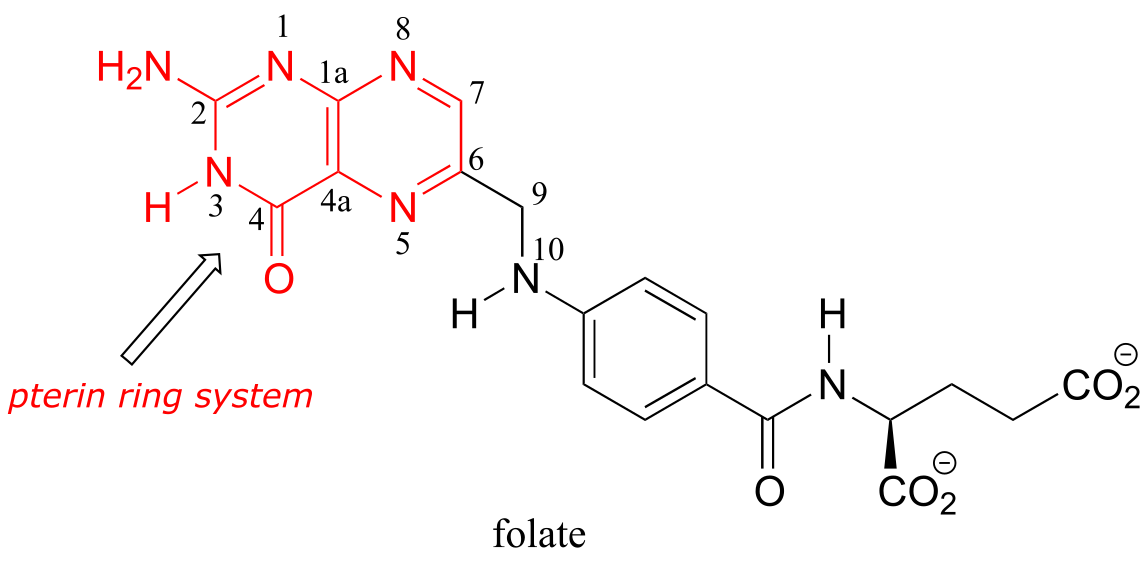
fig 50
17.4A: Active forms of folate#
Folate is active as a coenzyme in its reduced forms, dihydrofolate and tetrahydrofolate, which are formed by NADPH-dependent imine hydrogenation steps (section 15.3).
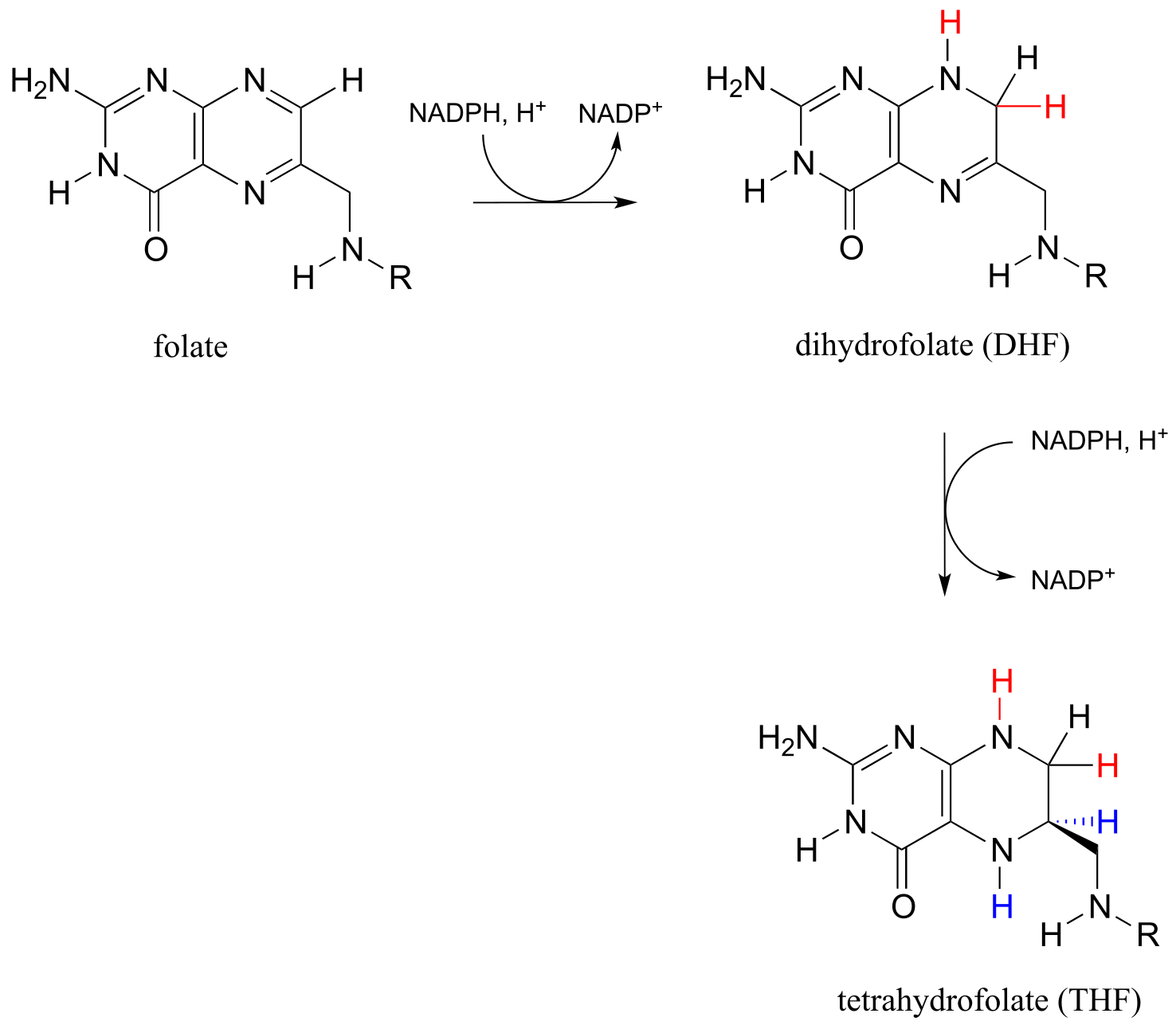
fig 51
The metabolic role of folate is to serve as a donor or acceptor in single-carbon transfer reactions. How is folate different from S-adenosylmethionine (SAM, section 8.8A) which also serves as a single-carbon donor? Recall that SAM donates a single carbon in the form of a methyl group: essentially, the single carbon of SAM is in the methanol (CH3OH) oxidation state, because it has only one bond to a heteroatom (specifically, to sulfur). (Refer to section 15.1 for a review of oxidation states).

fig 52a
Folate coenzymes, on the other hand, can carry a single carbon in the formaldehyde and formate oxidation states, in addition to the methanol oxidation state. By ‘formaldehyde’ and ‘formate’ oxidation state, we mean that the carbon has two and three bonds to heteroatoms, respectively.
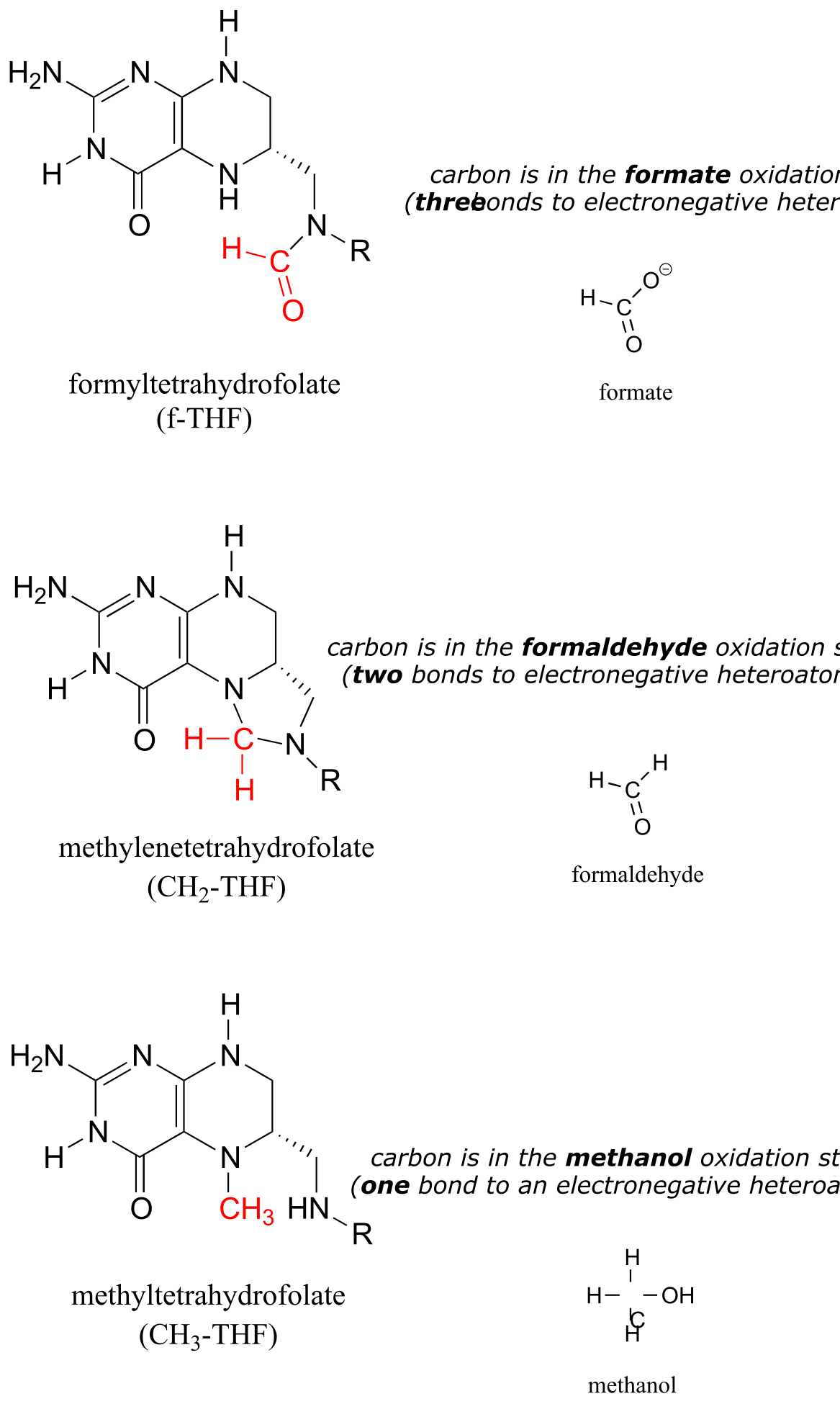
fig 52
Some key reactions in nucleic acid and amino acid metabolic pathways involve transfer of a single carbon in the formaldehyde or formate states. However, this could present problems. Formaldehyde by itself is very toxic: in particular, it tends to spontaneously form unwanted crosslinks between amine groups (eg. lysine side chains) in proteins.
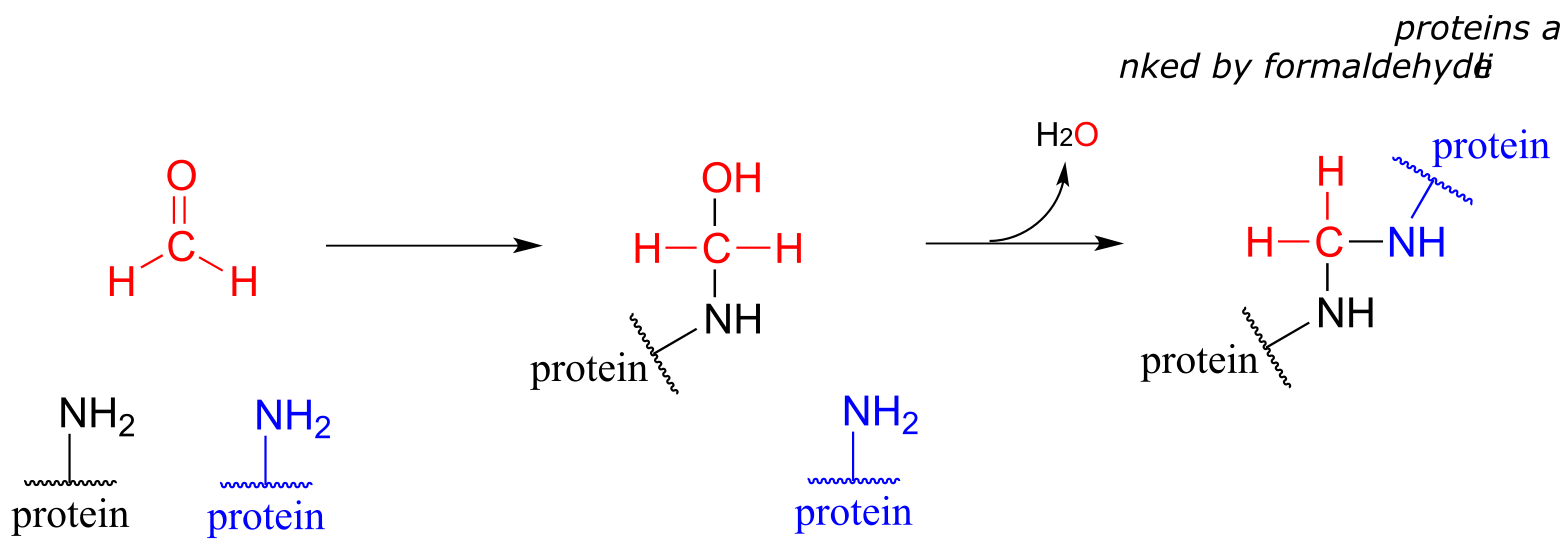
fig 53
Free formaldehyde is too reactive, and would cause damage to a cell. The CH2-THF coenzyme is stable in solution, but in the active site of certain enzymes it is reactive enough to serve as a formaldehyde donor, as we will see shortly.
Free formate, on the other hand, is a carboxylate, and we know from chapter 11 that carboxylates are not reactive in acyl substitution steps. Formate could be activated by phosphorylation, of course, but the resulting formyl phosphate would be too reactive in many enzyme active sites. A ‘happy medium’ has been found in which carbons in the formate oxidation state are carried by folate in the form of f-THF: once again, the carbon donor is stable in solution, but sufficiently reactive in certain enzyme active sites to accomplish controlled transfer of a formate group.
17.4B: Formation of formyl-THF and methylene-THF#
Formyltetrahydrofolate (f-THF) is formed from THF and a formate molecule which has been activated by phosphorylation (formyl phosphate, as stated in the paragraph above, is a high reactive intermediate, but is held inside the enzyme’s active site for immediate reaction with the incoming amine group of THF).
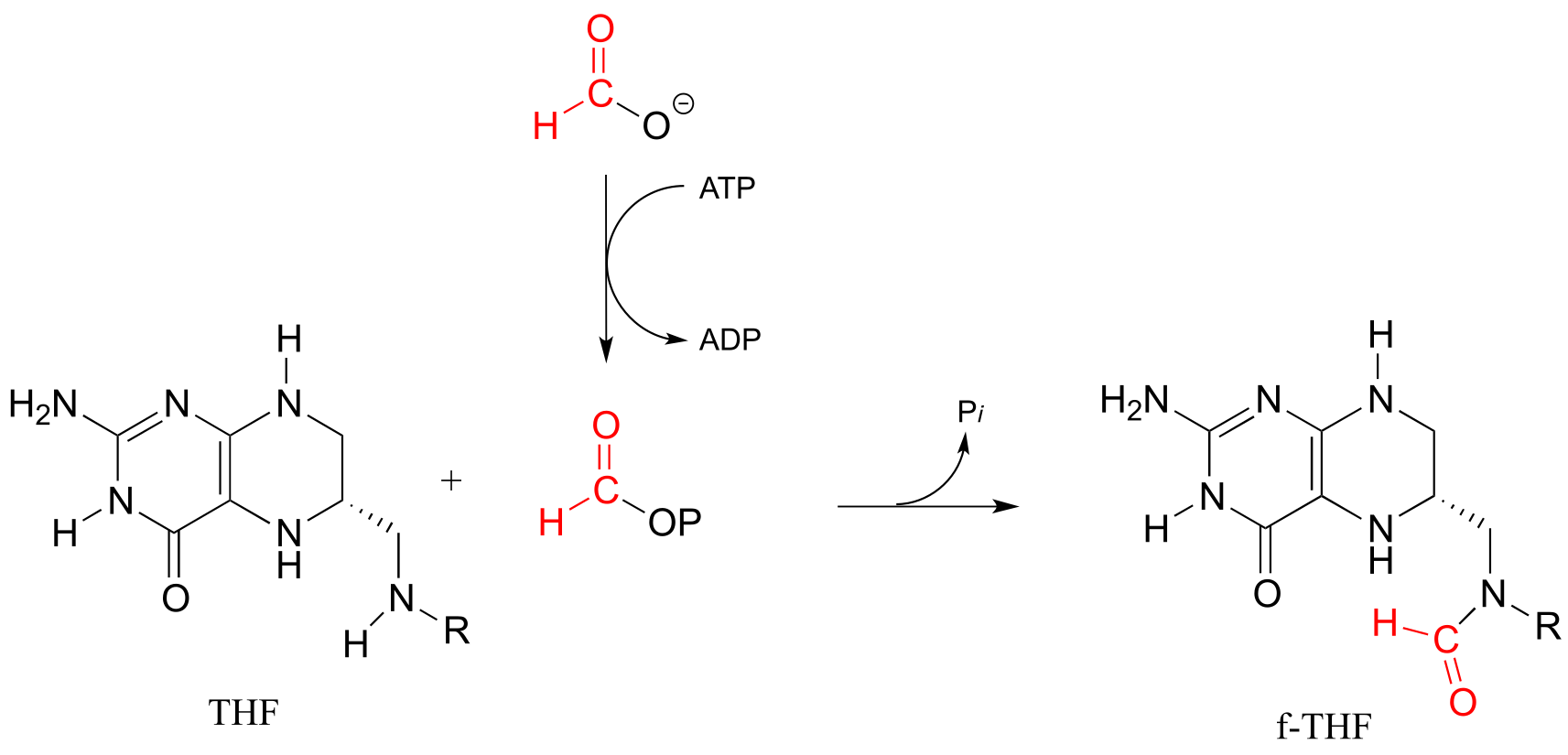
fig 54
There are two main metabolic routes to CH2-THF. One route is just the last step of the serine hydroxymethyltransferase reaction we have already seen in section 17.1D: the formaldehyde formed in the PLP-dependent phase of the reaction stays in the active site, and the oxygen is displaced by successive attacks from the amine nucleophiles at the 5 and 10 positions of THF. (Notice the similarity to the formaldehyde-protein crosslinking reaction shown earlier in this section.)
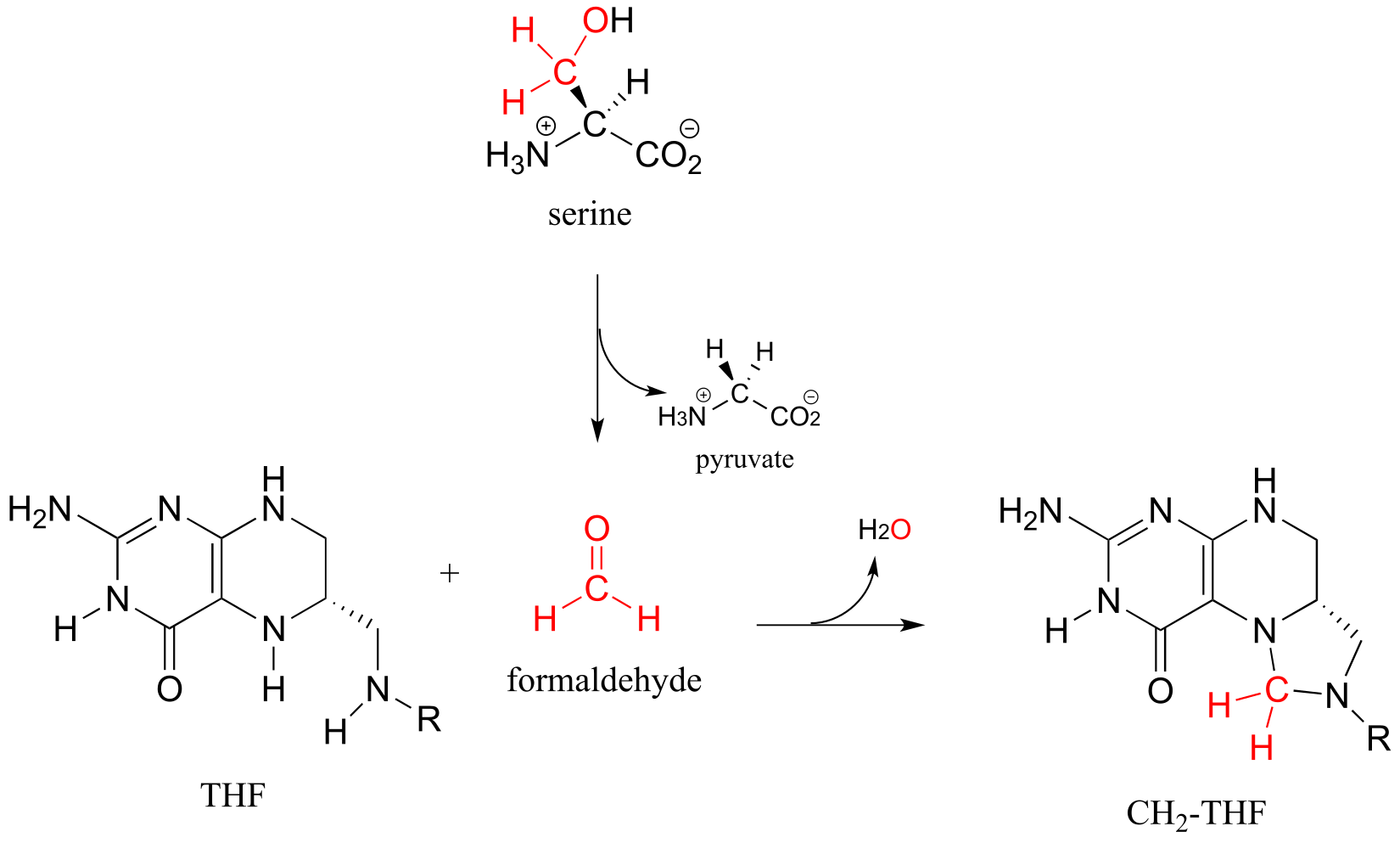
fig 55
In the second route, f-THF is dehydrated, then the resulting methenyl-THF intermediate is reduced by NADPH.
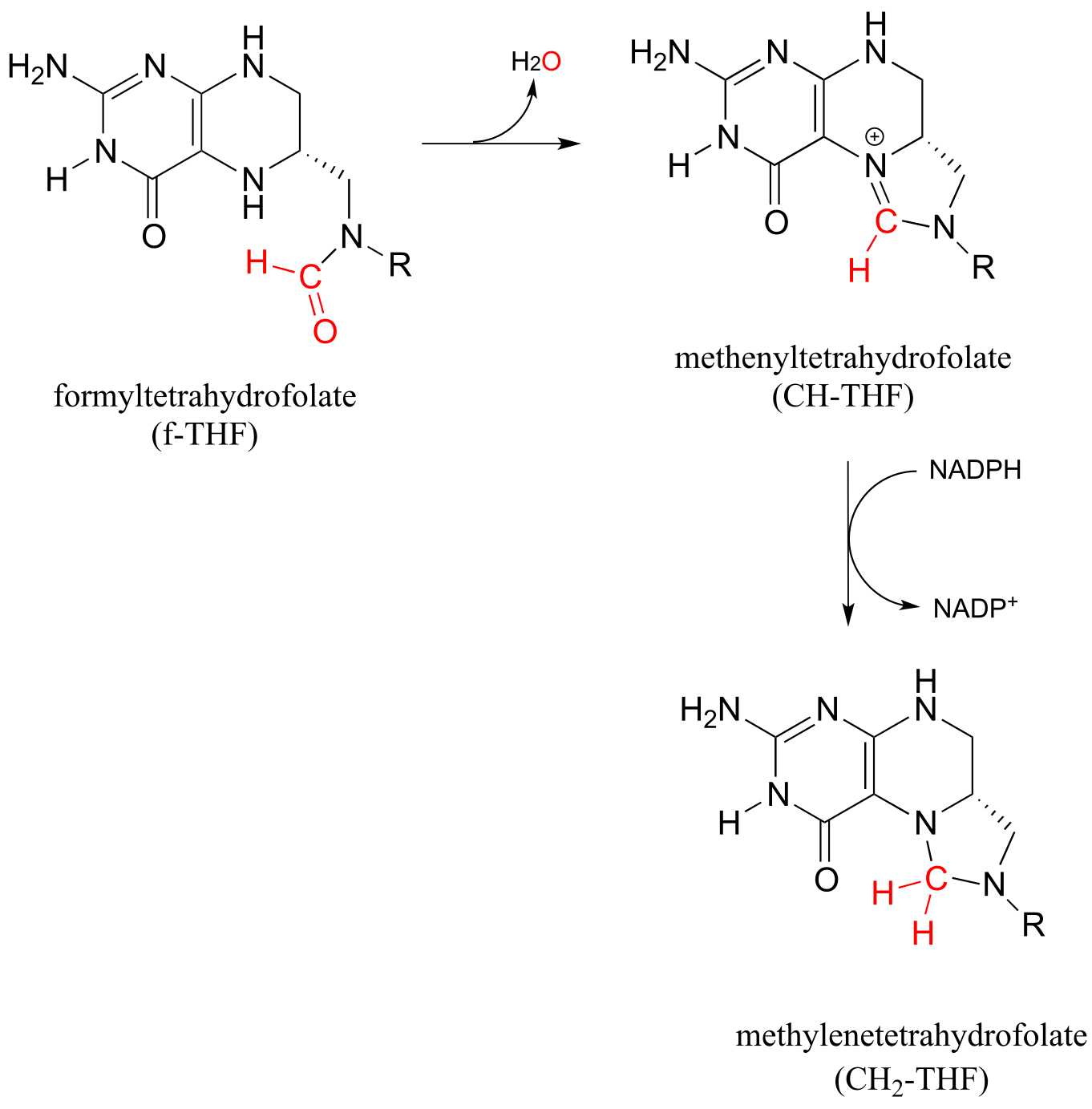
fig 56
Methylene-THF (CH2-THF) is reduced to methyl-THF (CH3-THF) in a flavin-dependent reaction. Biochemistry 2001, 40, 6216
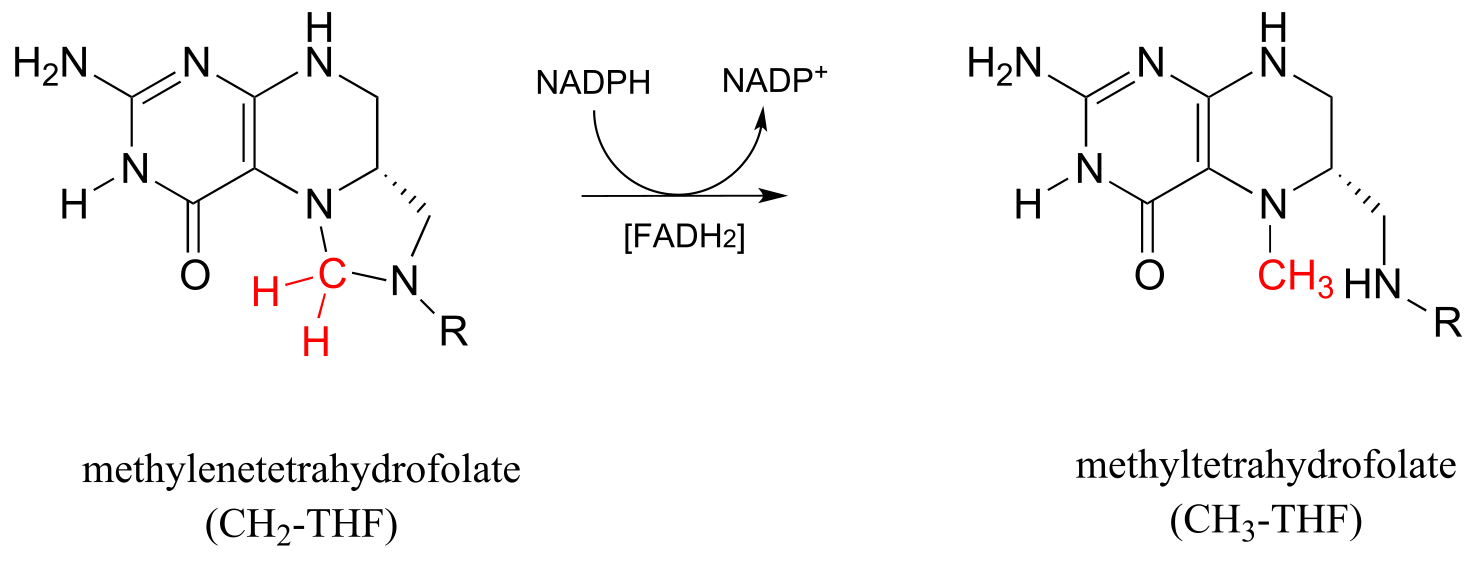
fig 56a
Recall from the introduction to this chapter that babies who were in the womb during the Dutch Hunger Winter faced a higher risk, when they reached adulthood, of conditions such as obesity and schizophrenia, most likely caused by disruptions in the S-adenosylmethionine-dependent methylation of their DNA, which was in turn caused by their mothers not getting enough folate. CH3-THF is the source of the methyl group in methionine, which ultimately becomes the methyl group in SAM. The methylation of homocysteine to methionine (below) involves a cobalt-containing coenzyme called cobalamin, but the mechanism for this reaction is beyond the scope of our discussion. The second reaction below (formation of SAM) is simply an SN2 displacement of the inorganic triphosphate leaving group on ATP by the nucleophilic sulfur in methionine.
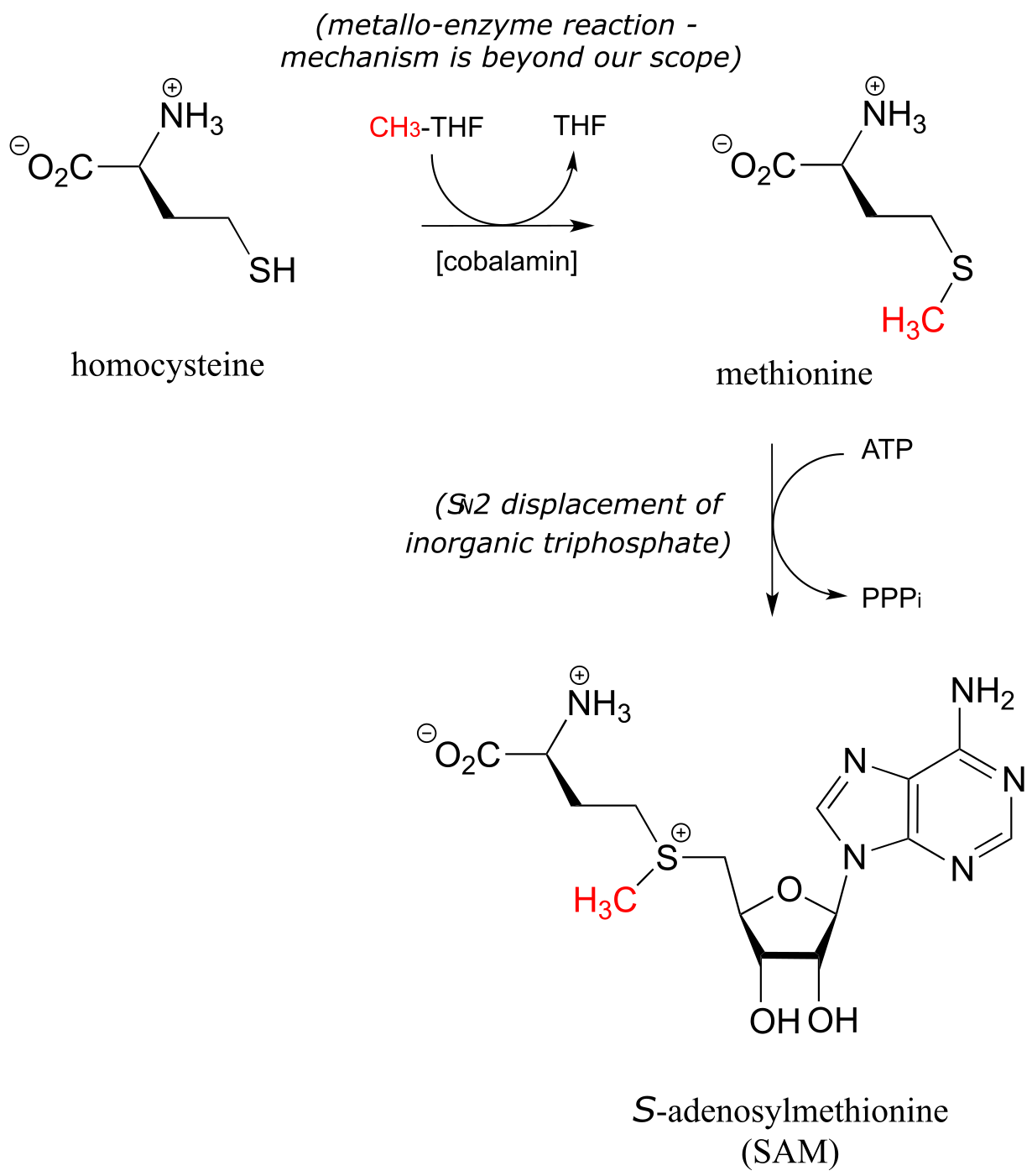
fig 56b
17.4C: Single-carbon transfer with formyl-THF#
There are two important f-THF-dependent formylation steps in the biosynthetic pathways for purine nucleophiles. Both are simply transamidation reactions: in other words, conversion by the nucleophilic acyl substitution mechanism (chapter 11) of one amide to another.
Glycinamide ribonucleotide transformylase reaction:
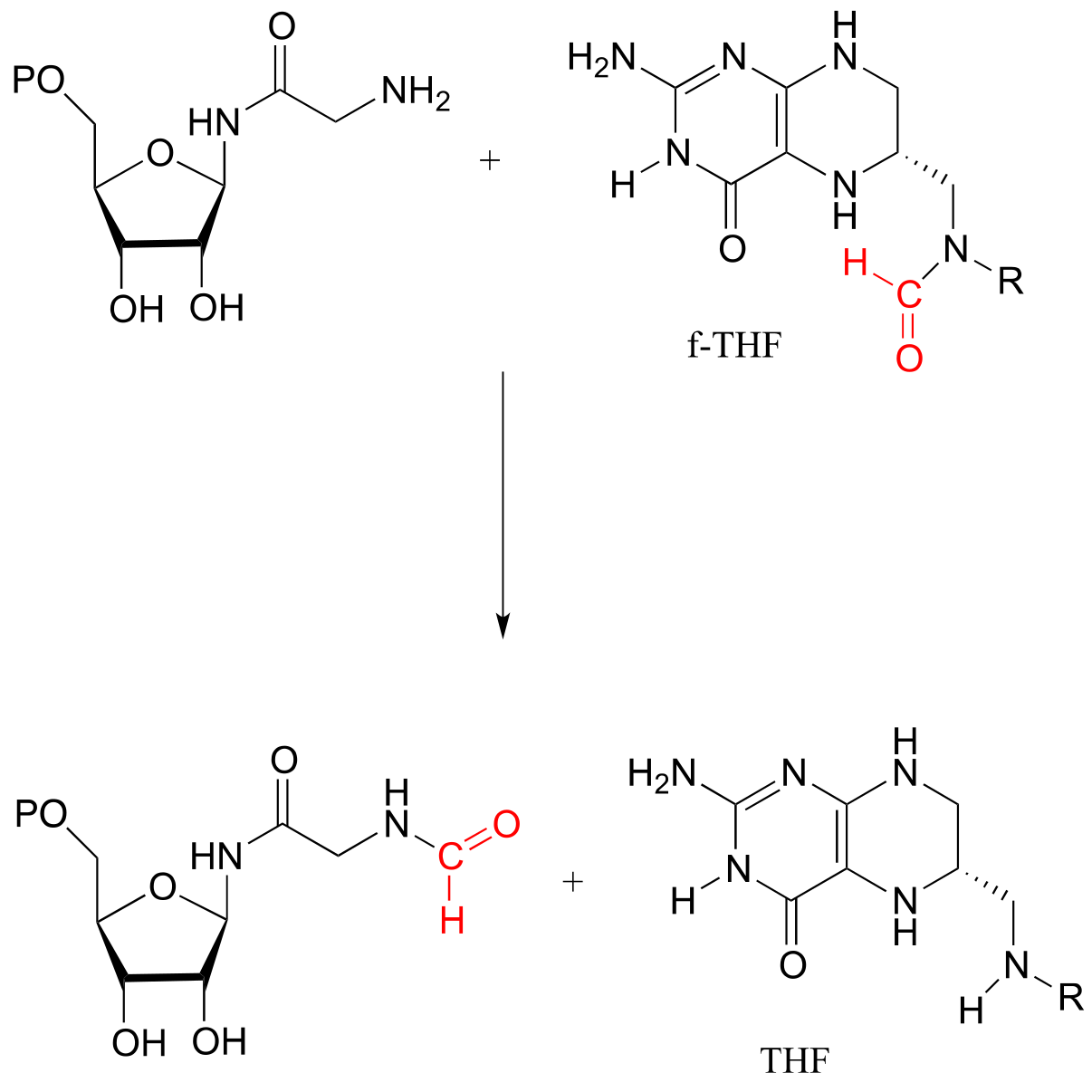
fig 57
Aminoimidazole carboxamide transformylase reaction:
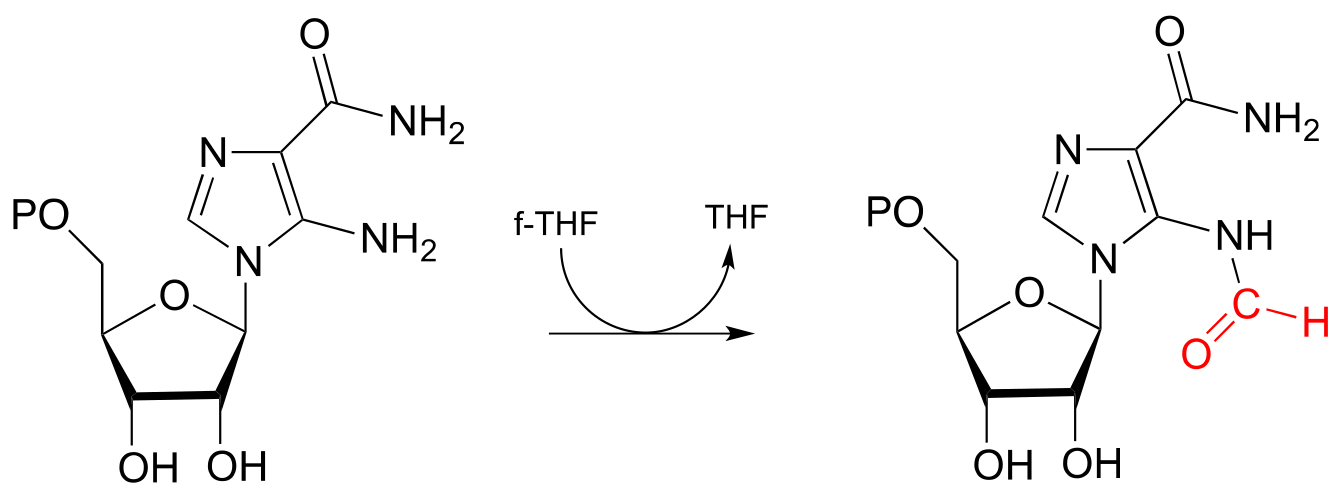
fig 58
Exercise 17.9: Which of the two transformylase reactions above would you expect to be more kinetically favorable? Explain your reasoning.
Exercise 17.10: Predict the structure of ‘product X’ in the reaction below, from the histidine degradation pathway.
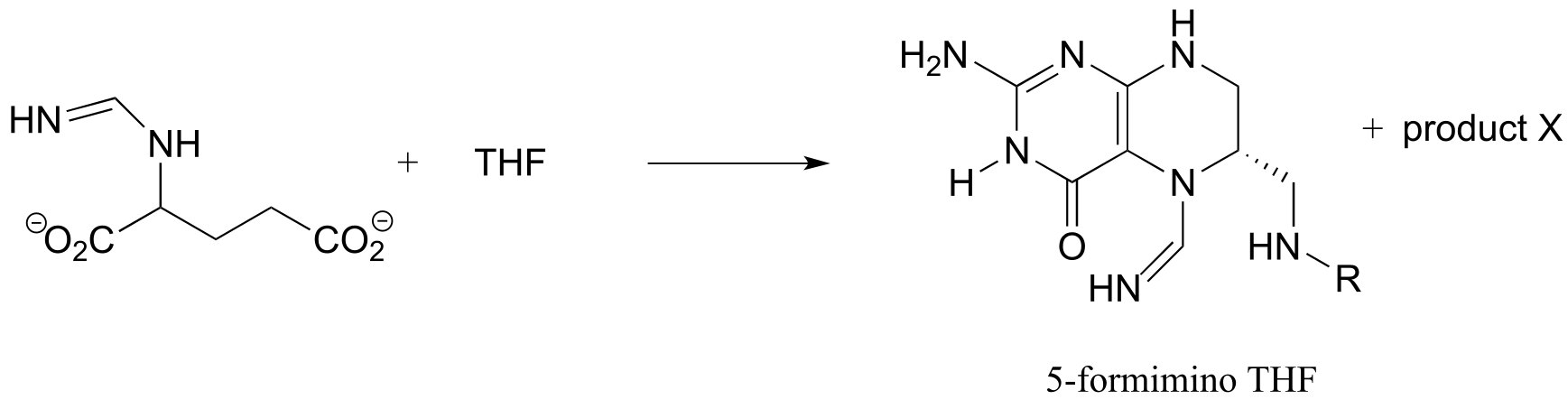
fig 59
17.4D: Single-carbon transfer with methylene-THF#
CH2-THF serves as a single-carbon donor in a somewhat complicated reaction in the biosynthesis of the DNA monomer doexythymidine monophosphate (dTMP).
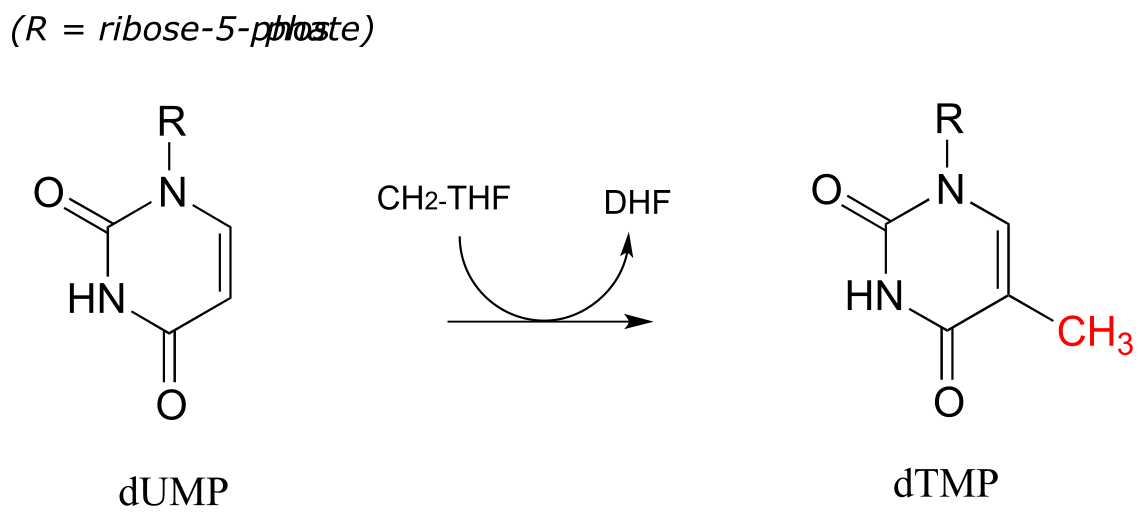
fig 60
The reactions begins with the five-membered ring of CH2-THF breaking apart to create an imine intermediate (step 1 below). The imine becomes an electrophile in a conjugate addition step (section 13.4, steps 2-3 below). Note that after step 1, the second ring in the pterin system of the coenzyme is abbreviated for clarity.
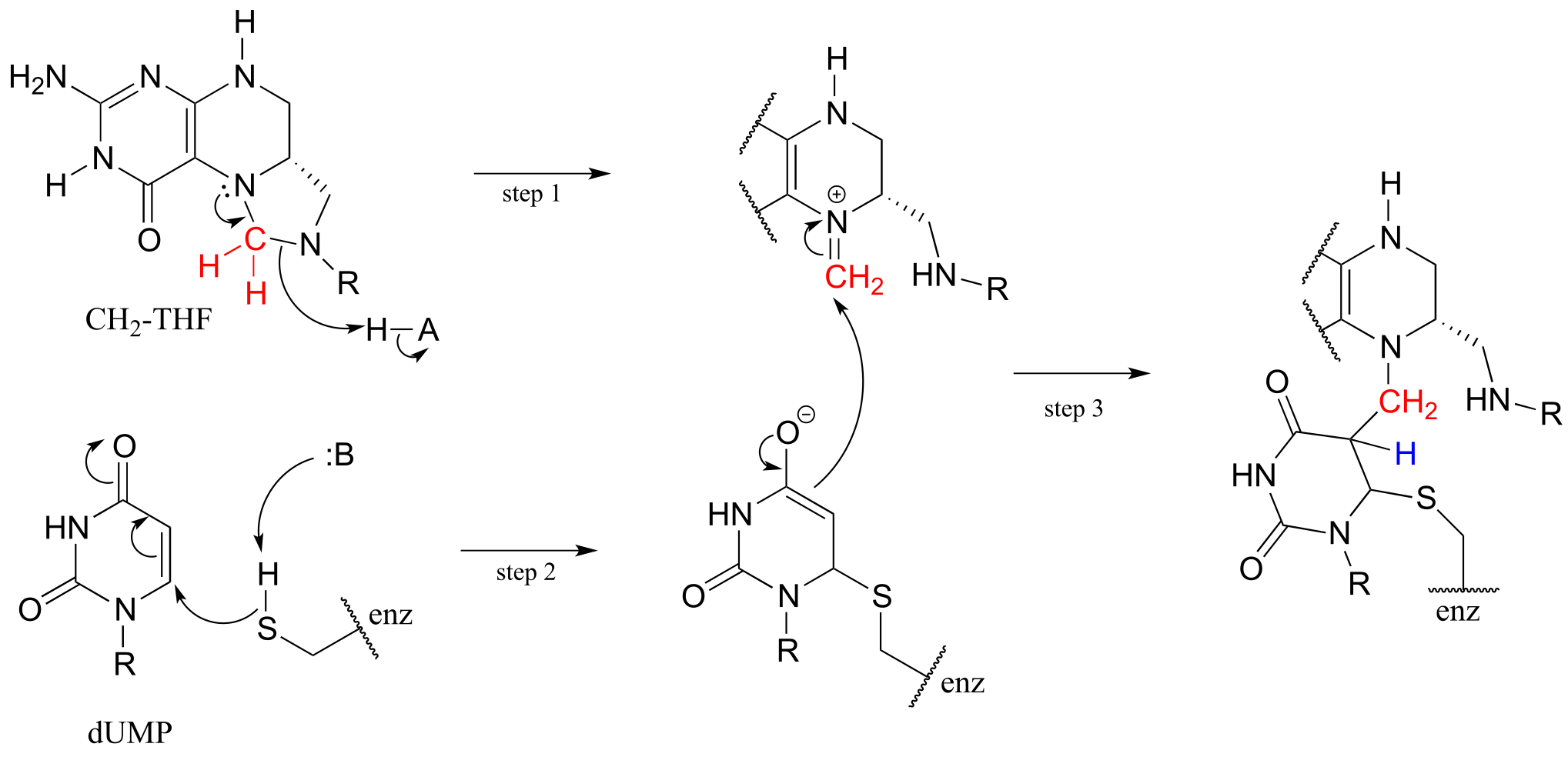
fig 61
Next, tetrahydrofolate is eliminated in an E1cb elimination mechanism (steps 4 and 5). Notice that this is where the single carbon is transferred from methyl-THF to dUMP.
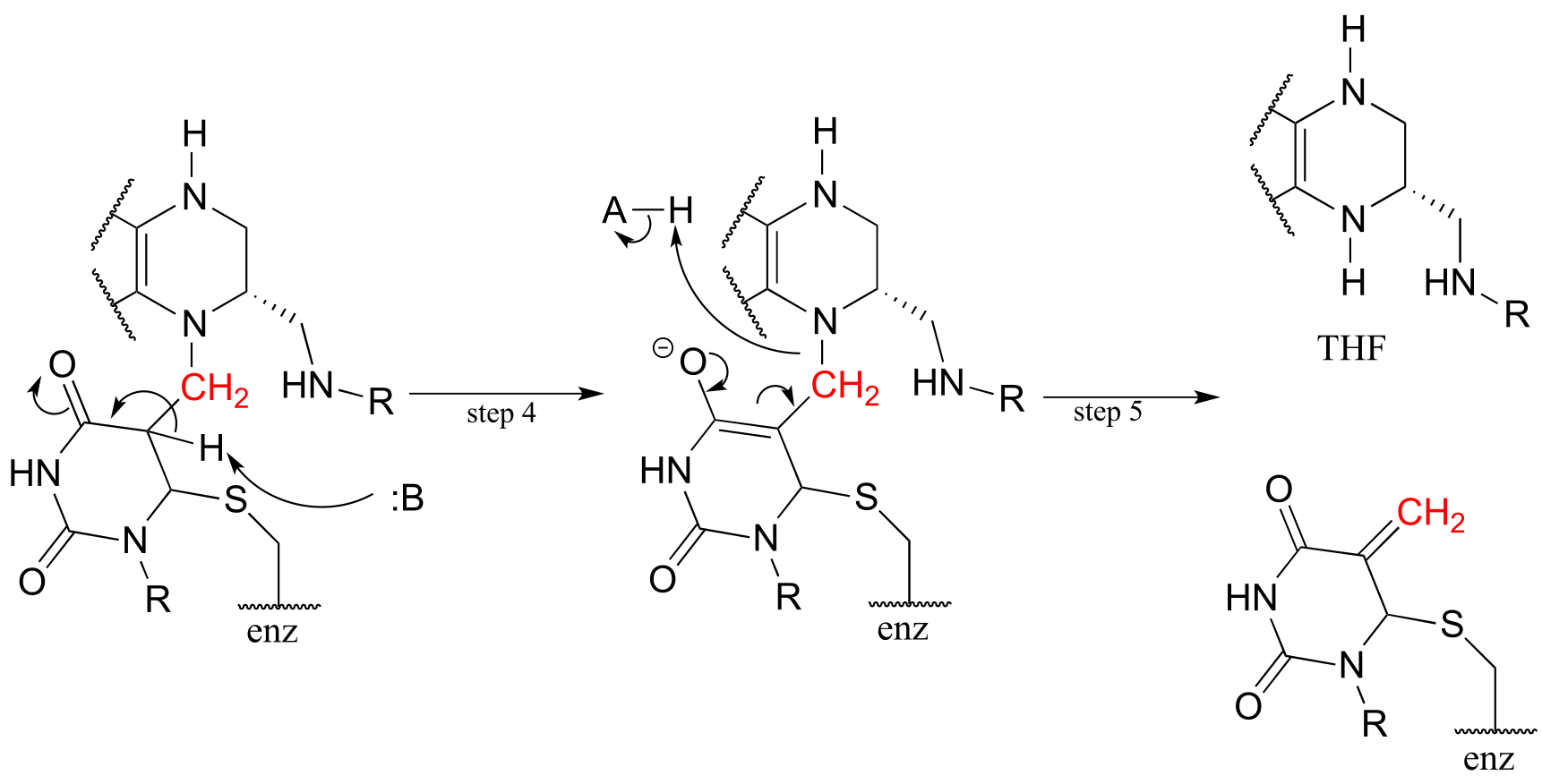
fig 62
The final step in the mechanism is where it gets really interesting: a hydride ion is transferred from the tetrahydrofolate coenzyme to the methylene (CH2) group on the deoxynucleotide substrate. Essentially, this is a conjugated SN2 step with hydride as the nucleophile and the active site cysteine as the leaving group.
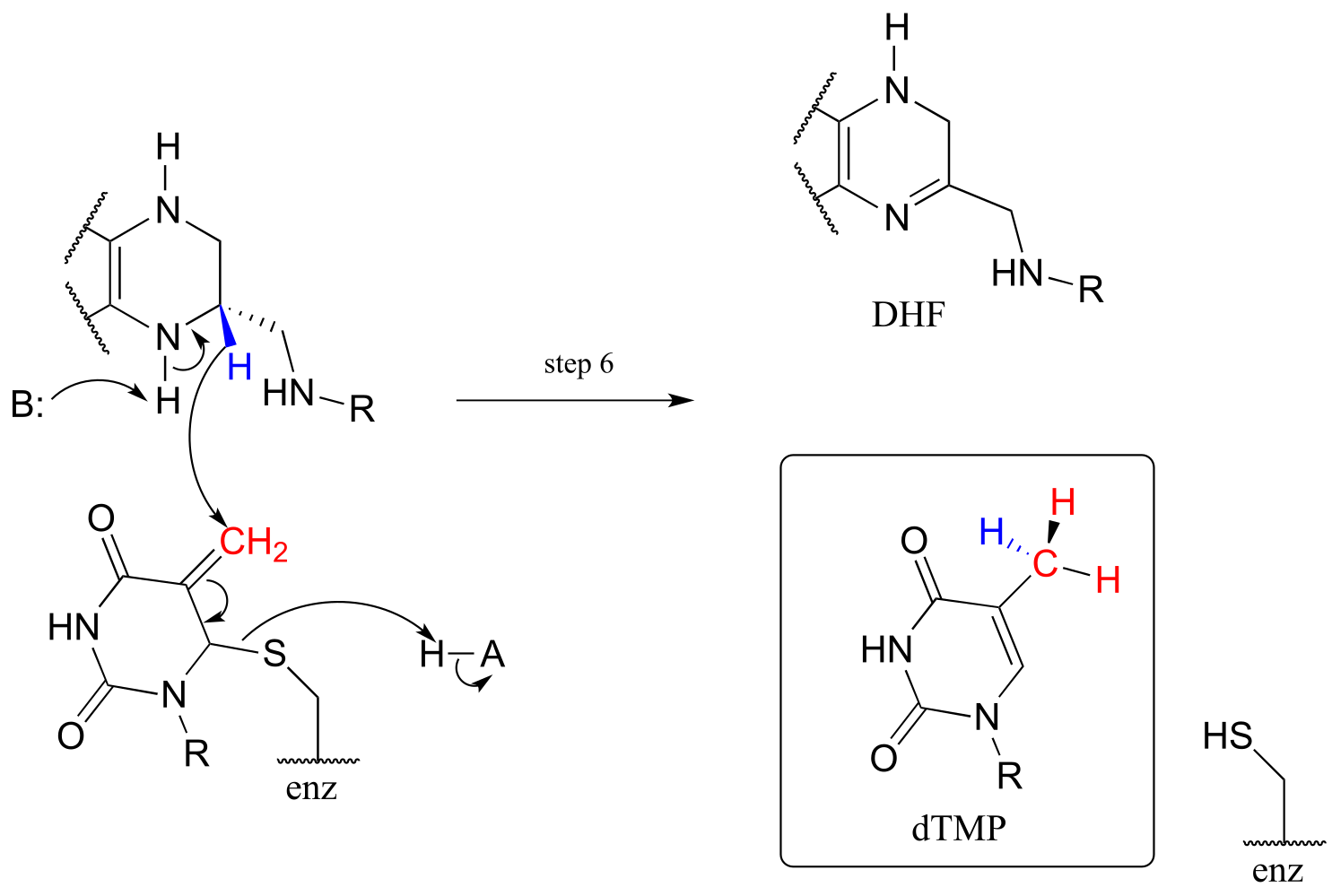
fig 63
**
**
Key learning objectives for this chapter#
After completing this chapter, you should be able to:
Understand how pyridoxal phosphate (PLP) acts as an ‘electron sink’ in a variety of reactions in amino acid metabolism.
Recognize and draw mechanisms for PLP-dependent transformations of the following types:
racemization
decarboxylation
transamination
retroaldol cleavage
retro-Claisen cleavage
β-elimination
β-substitution
γ-elimination
γ-substitution
Recognize transformations - amino acid decarboxylation and transamination, for example - in which chemical steps occur that simply don’t ‘make sense’ unless the electron sink role of PLP is taken into account.
Understand how the orientation of the substrate in relation to the plane formed by the conjugated π system of PLP is a major factor in catalysis of PLP-dependent reactions.
Understand how thiamine diphosphate (ThDP) acts as an ‘electron sink’ in a variety of reactions in which a bond to a carbonyl carbon is broken, and how these steps do not ‘make sense’ unless the electron sink role of ThDP is taken into account.
Recognize transformations for which ThDP is likely required, and be able to draw reasonable mechanisms for them.
Understand how ThDP acts in tandem with lipoamide, flavin, and nicotinamide in the reaction catalyzed by pyruvate dehydrogenase.
Recognize folate in its various forms - DHF, THF, f-THF, CH2-THF, and CH3-THF - functions in a variety of one-carbon transfer steps. Be able to recognize the oxidation state of the carbon being transferred in a folate-depenent step.
Problems#
P17.1: Here is a chance to test your ability to recognize reactions catalyzed by enzymes using three coenzymes - thiamine diphosphate, pyridoxal phosphate, and folate - that we have studied in this chapter. For each generalized reaction, look carefully at the transformation that is taking place, and decide which of the three coenzymes is likely to be required. Then, draw the single mechanistic step by which the bond identified by an arrow is broken or formed. In the cases where a double bond is indicated, show the mechanistic step in which the σ bond is formed. In each case, your drawing should include the structure of the reactive part of the coenzyme, and should clearly show the role it plays in catalyzing the mechanistic step you are drawing.
a)
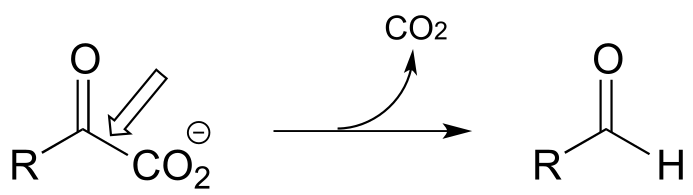
b)
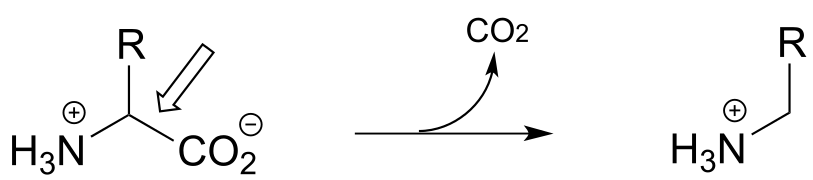
c)
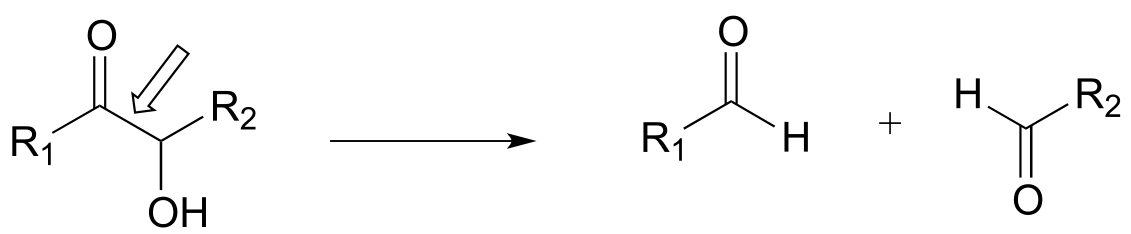
d)
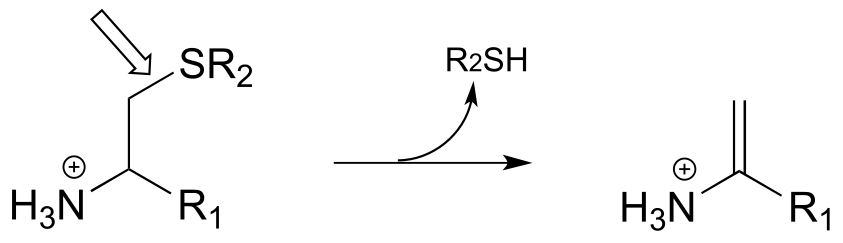
e)
f)
g)
h)
i)
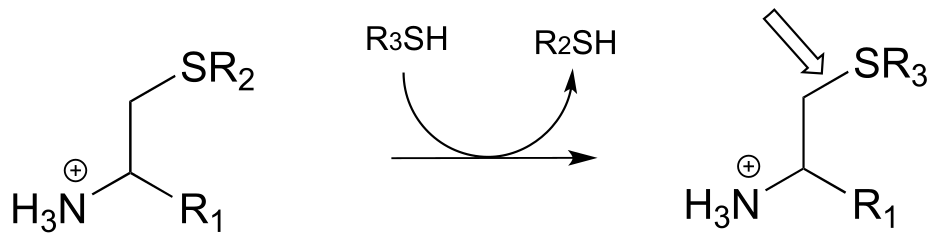
j)
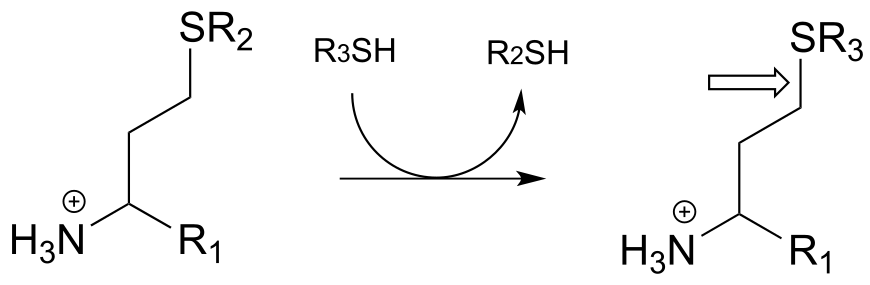
k)
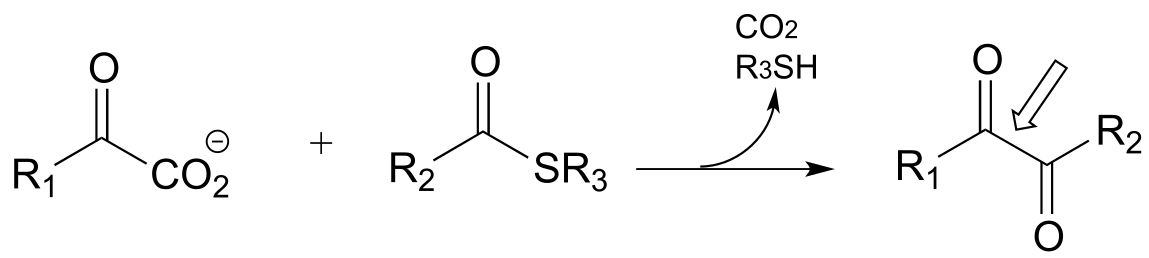
l)

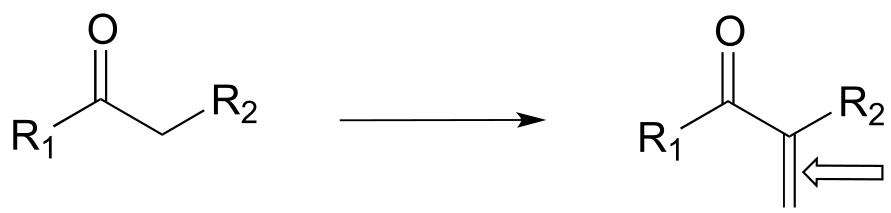
m)
P17.2: The final step in the biosynthesis of the amino acid tryptophan is a PLP-dependent condensation between serine and indole, shown below (EC 4.2.1.20). The reaction mechanism involves steps that are familiar from this chapter, but also incorporates a reaction type we studied in chapter 14. Propose a mechanism.
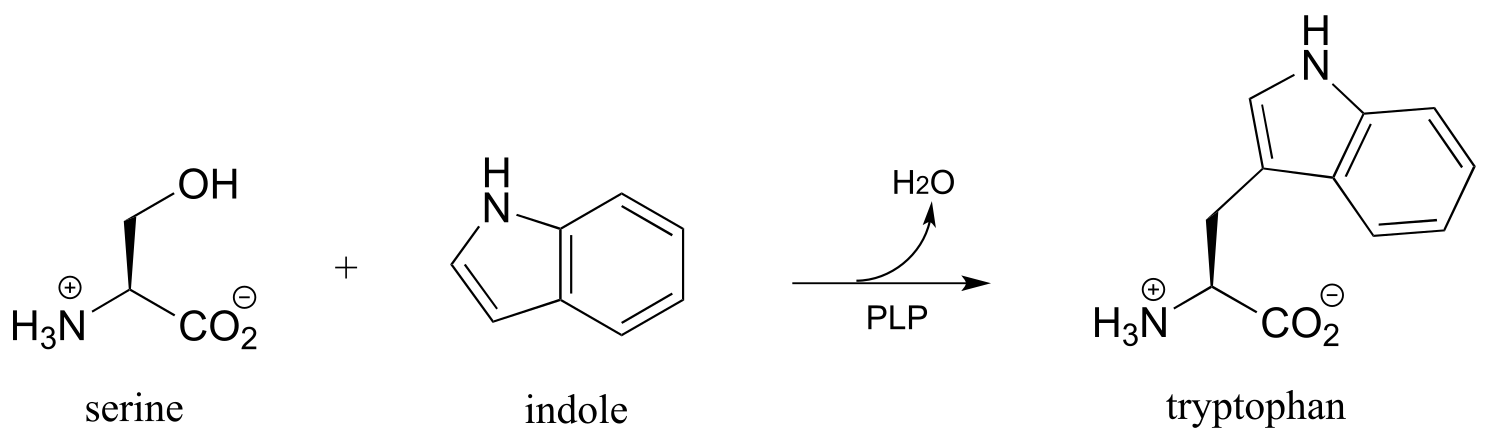
P17.3: Draw a reasonable mechanism for the following reaction, identifying the species denoted by questions marks. (Biochemistry 2012, 51, 3059)
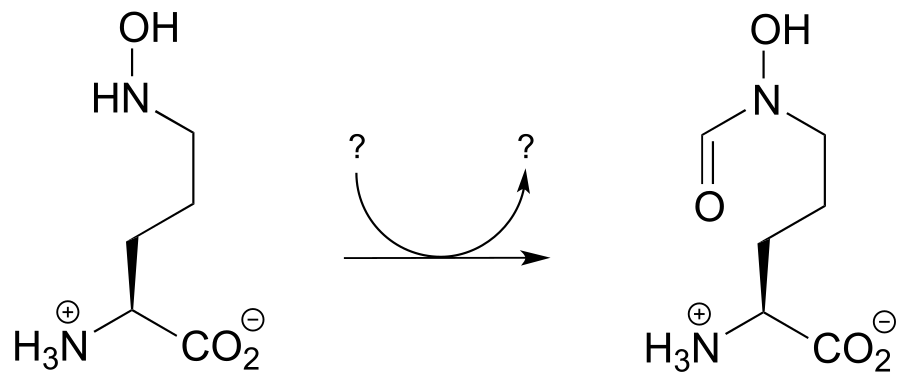

P17.4: Propose a mechanism for the reaction below, which is part of the anaerobic catabolism of alcohols in some species of bacteria. (ChemBioChem 2014, 15, 389)
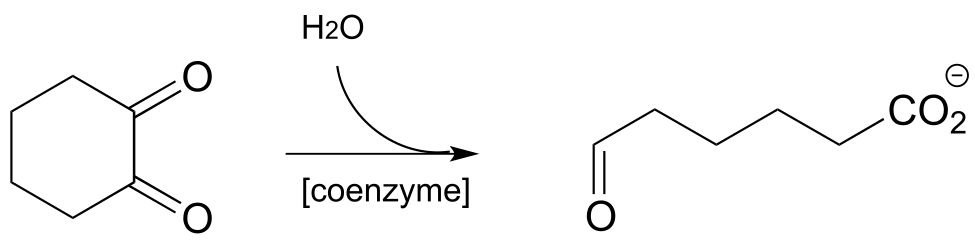
P17.5: Identify cosubstrate A and propose a mechanism for the reaction shown below, which was reported to occur in the thermophilic bacterium Thermosporothrix hazakensis. (ChemBioChem 2014 15, 527).
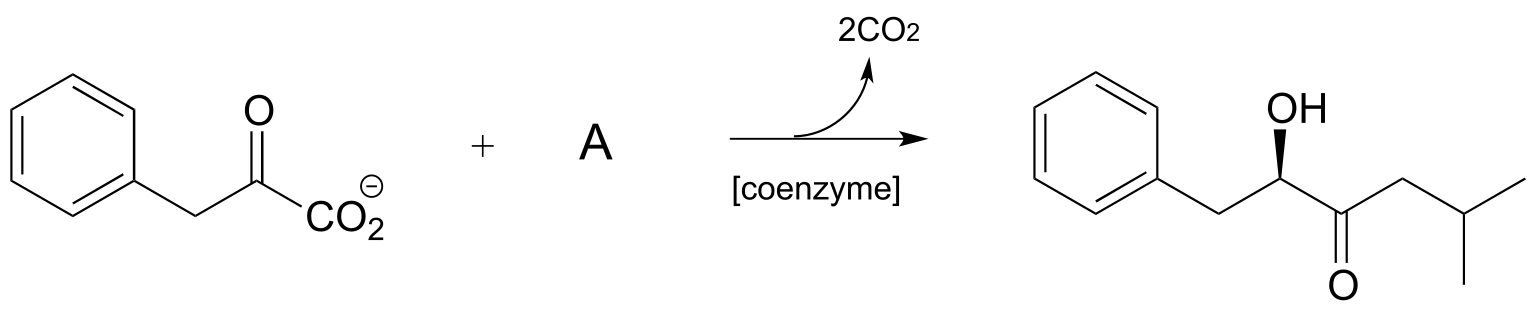
P17.6: Propose a mechanism for each of the reactions below, being sure to show the role played by the coenzyme (you need to determine which coenzyme is needed in each case).
a)
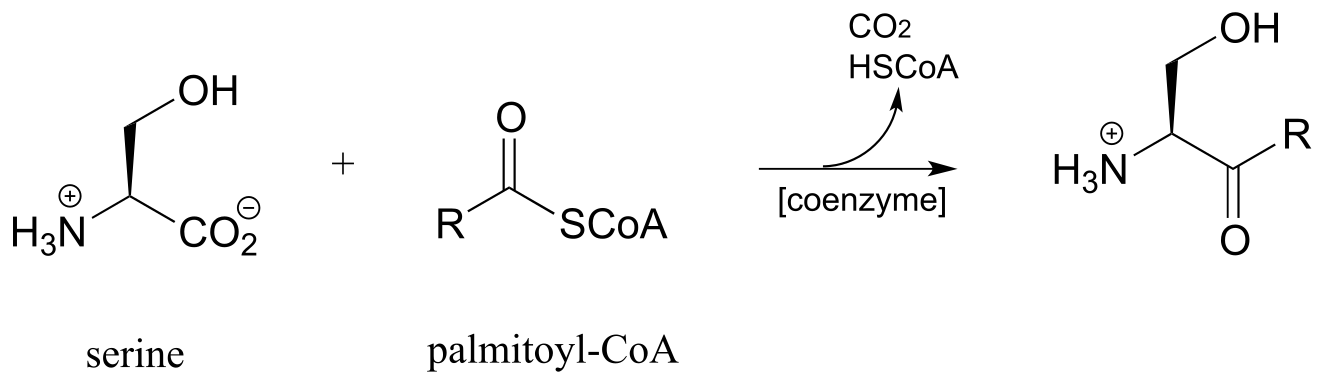
b) (E.C. 2.2.1.6)
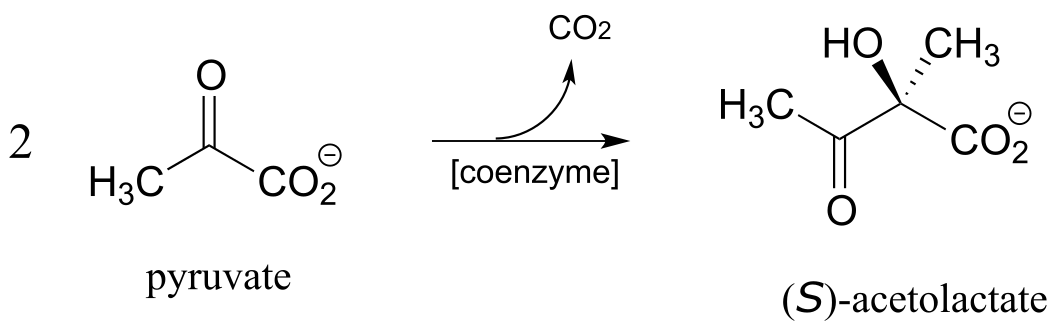
c) Angew. Chem. Int. Ed. Engl. 2014, 53, 1943.
http://onlinelibrary.wiley.com/doi/10.1002/anie.201307989/full Fig 1 Orf R rxn
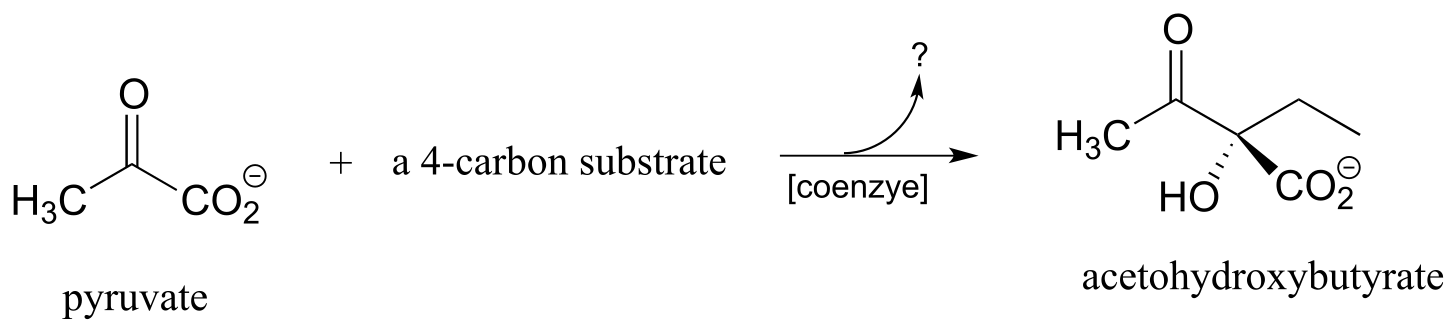
P17.7: Acetohydroxybutyrate is formed in a coenzyme-dependent reaction between pyruvate and a 4-carbon compound. What is a likely second substrate, coenzyme, and by-product (indicated below with a question mark)?
http://www.sciencedirect.com/science/article/pii/S1367593105001043
P17.8:
a) The ‘benzoin condensation’ reaction was discovered in the 19th century, and led eventually to a better understanding of ThDP-dependent reactions in the cell. In a traditional benzoin condensation reaction, cyanide ion (instead of ThDP) plays the role of electron acceptor. Enzyme-catalyzed benzoin condensation reactions are also known to occur in some bacteria*:* Pseudomonas fluorescens, for example, contains an enzyme that catalyzes the synthesis of (R)-benzoin.
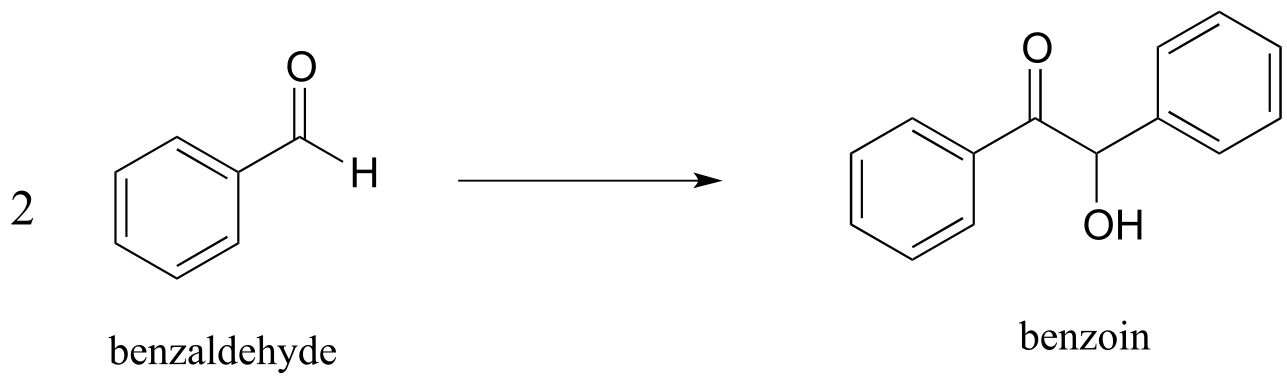
a) Draw a mechanism for the enzyme-catalyzed (ThDP-dependent) benzoin condensation reaction*.*
b) Draw a mechanism for the cyanide-catalyzed benzoin condensation reaction (non-enzymatic, basic conditions).
c) The following ThDP-reaction was recently reported to be part of the biosynthetic pathway for clavulanic acid, a compound that inhibits the action of β-lactamases (β-lactamases are bacterial enzymes that hydrolyze penicillin-based antibiotic drugs, rendering them ineffective). As is typical for ThDP-dependent reactions, the first step is addition of the ylide form of the coenzyme to the substrate carbonyl. The next steps are (in order): dehydration, tautomerization, elimination of phosphate, conjugate addition of arginine, and finally hydrolytic cleavage of the coenzyme-product bond. Draw out a complete mechanism that corresponds to this description.
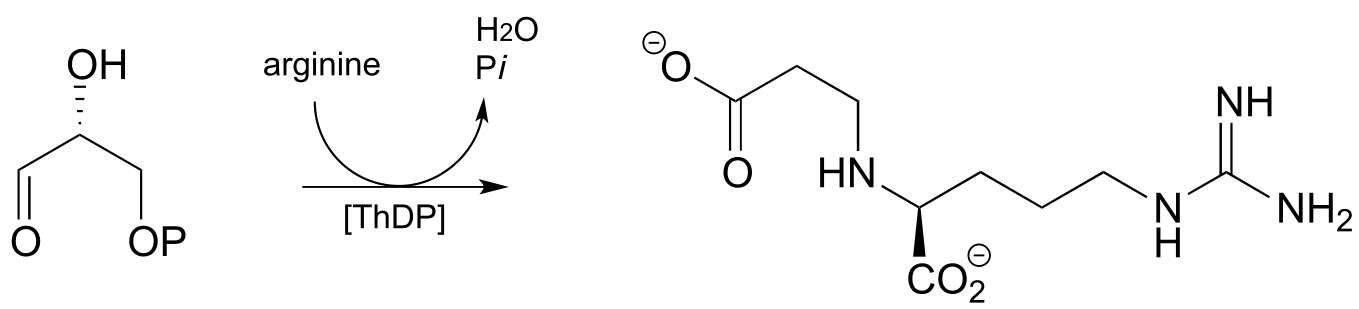
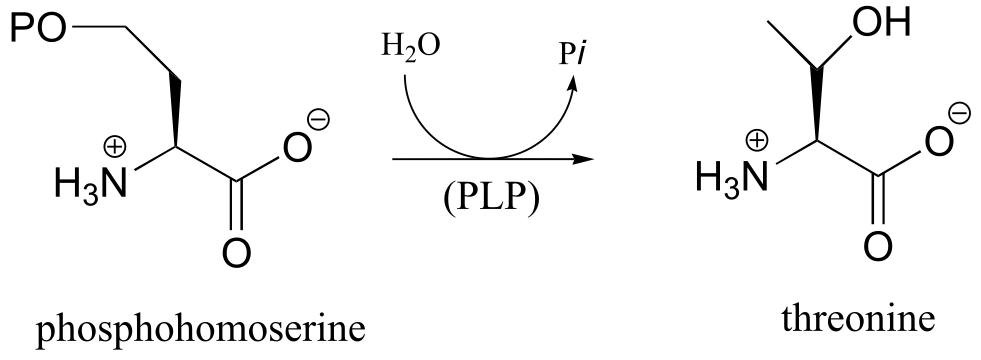
P17.9: Practice with PLP-dependent reactions:
a) Propose a mechanism for this reaction, which is part of the tryptophan degradation pathway (EC 3.7.1.3).
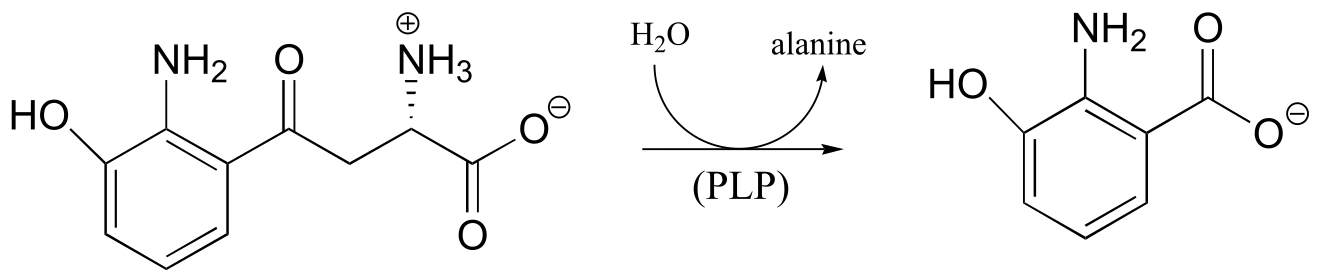
b) Propose a mechanism for the final step of the threonine biosynthesis pathway (EC 4.2.3.1).
c) Propose a mechanism for the reaction catalyzed by aspartate β-decarboxylase (EC 4.1.1.12), which converts aspartate to alanine in a PLP-dependent reaction.
d) Sphingolipids are a type of membrane lipid found in the membranes of all eukaryotic cells, and are most abundant in the cells of central the central nervous system. Synthesis of sphingolipids involves the PLP-dependent reaction below, catalyzed by serine palmitoyl transferase (EC 2.3.1.50). Propose a mechanism.
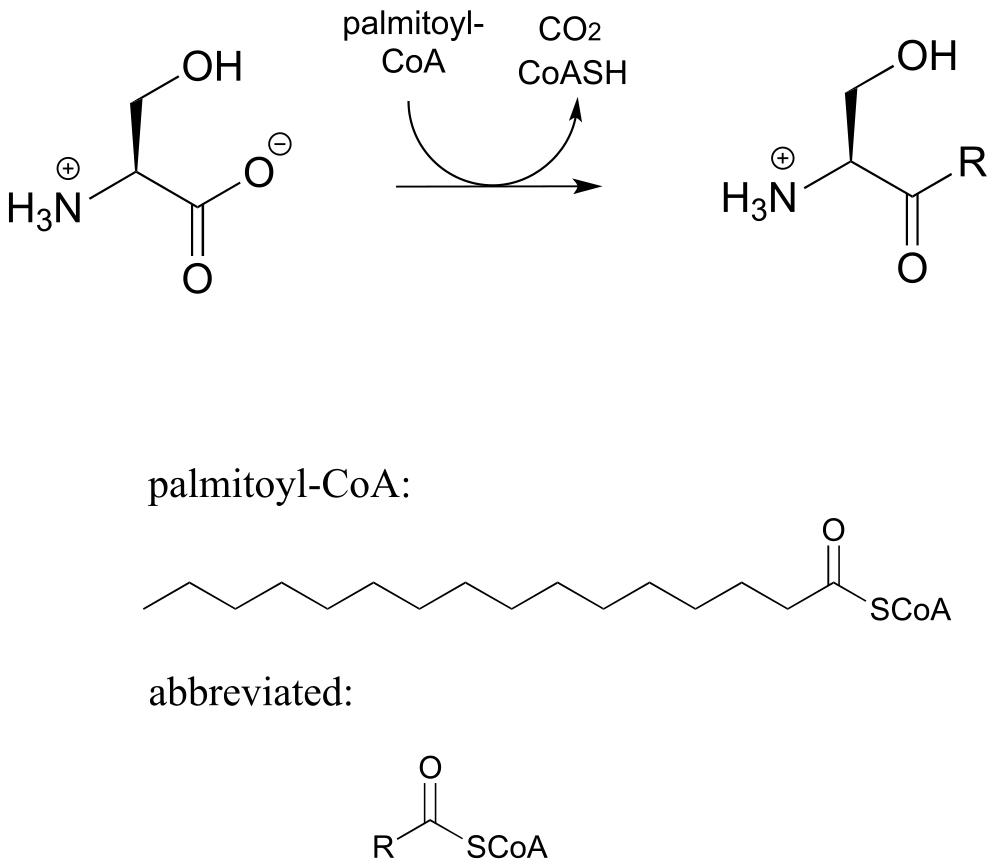
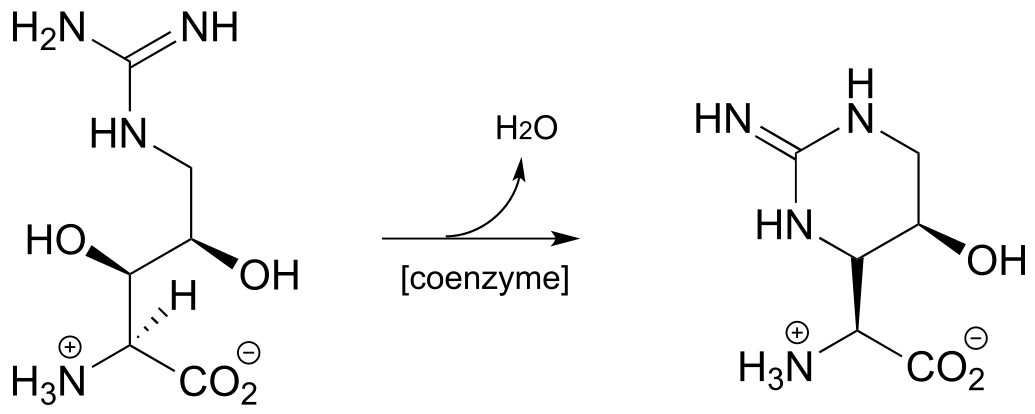
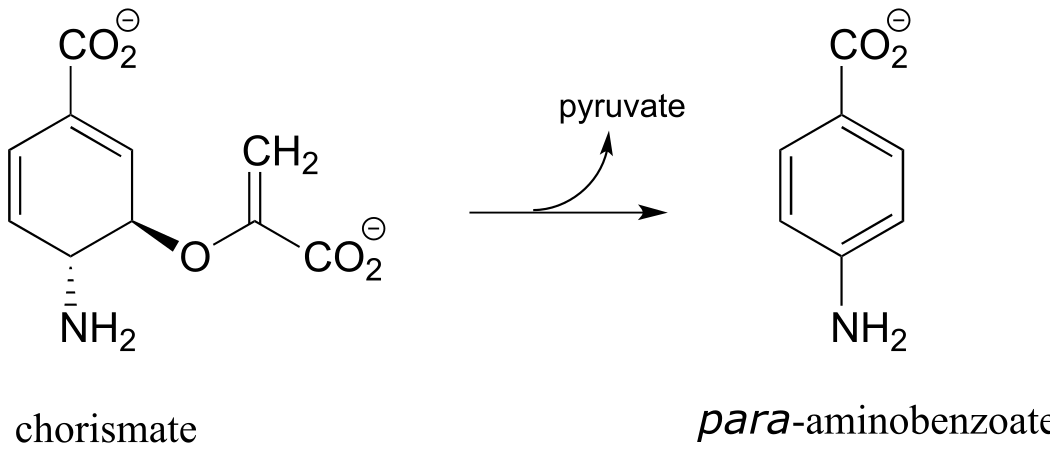
P17.10: As we saw in this chapter, PLP-dependent enzymes usually catalyze reactions involving amino acid substrates. Here is an exception, a PLP-dependent β-elimination reaction in the folate biosynthetic pathway (EC 4.1.3.38). Propose a mechanism for this reaction.
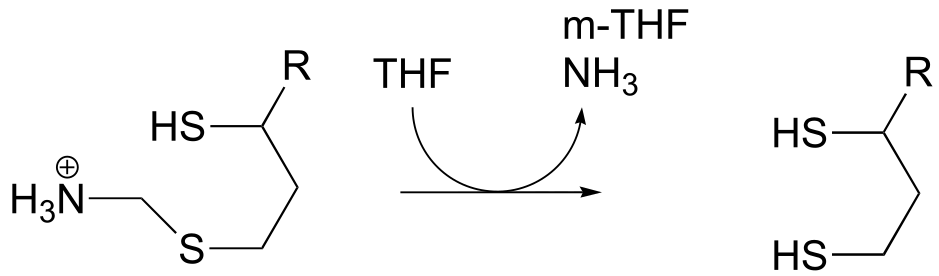
P17.11: The final step in the degradation pathway for the amino acid glycine (also known as the ‘glycine cleavage system’) is shown below. Propose a likely mechanism, given that
evidence suggests that CH2NH2+ is an intermediate.
P17.12: As we saw in chapter 15, the usual biochemical role of NAD+ is to act as a hydride acceptor in dehydrogenation reactions. An exception is the reaction catalyzed by the histidine degradation pathway enzyme urocanase (EC 4.2.1.49).
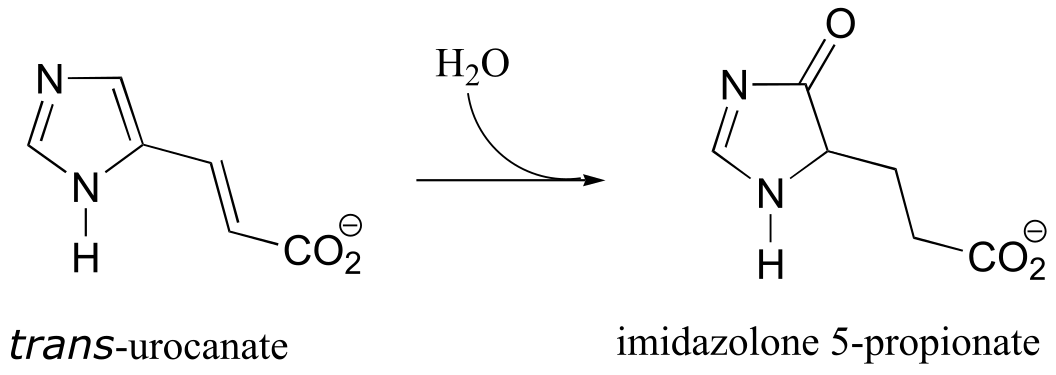
In this reaction, NAD+ acts as a catalytic, electron-sink coenzyme - it temporarily accepts electrons from a pi bond in the substrate, resulting in a covalent substrate-NAD adduct. This allows a key isomerization step to occur on the substrate through a protonation-deprotonation mechanism, followed by addition of water, cleavage of the substrate-NAD adduct to regenerate NAD+, and finally tautomerization to the product. Propose a mechanism that fits this description, and involves the intermediate below.
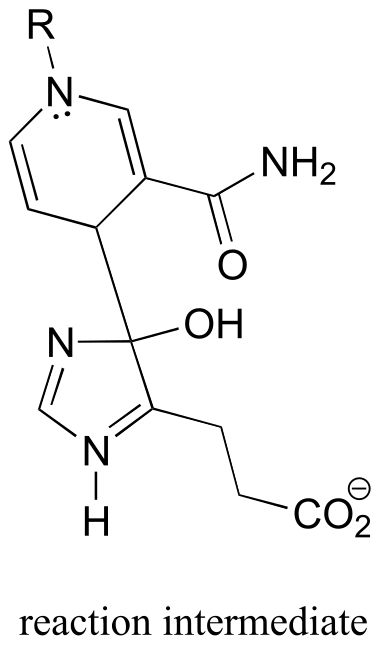
Pathway prediction problems#
**
**
P17.13: Propose a multistep pathway for each of the following
transformations. All involve at least one step requiring PLP, ThDP, or
folate.
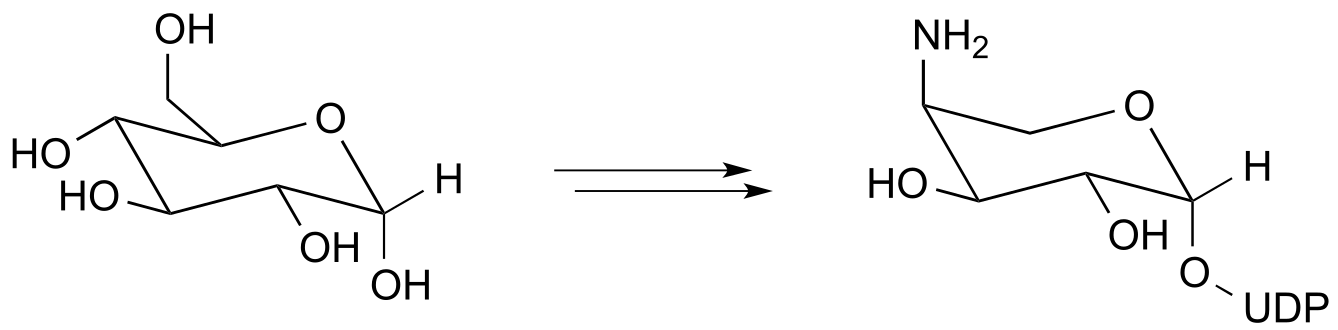
a) Below is portion of the biosynthesis of a modified membrane lipid in Salmonella and other pathogenic bacteria. The modified membrane confers antibiotic resistance to the bacterium. Biochemistry 2014, 53, 796
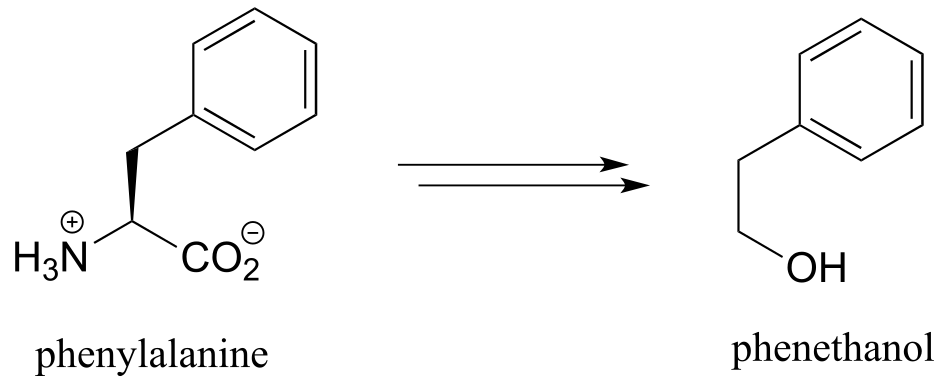
b) Below is the biosynthetic pathway for phenethanol in yeast. Phenethanol, which has a rose scent, is commonly used as a fragrance - this pathway has been proposed as a potential ‘green’ enzymatic synthesis to replace the traditional industrial synthesis, which uses toxic reagents.
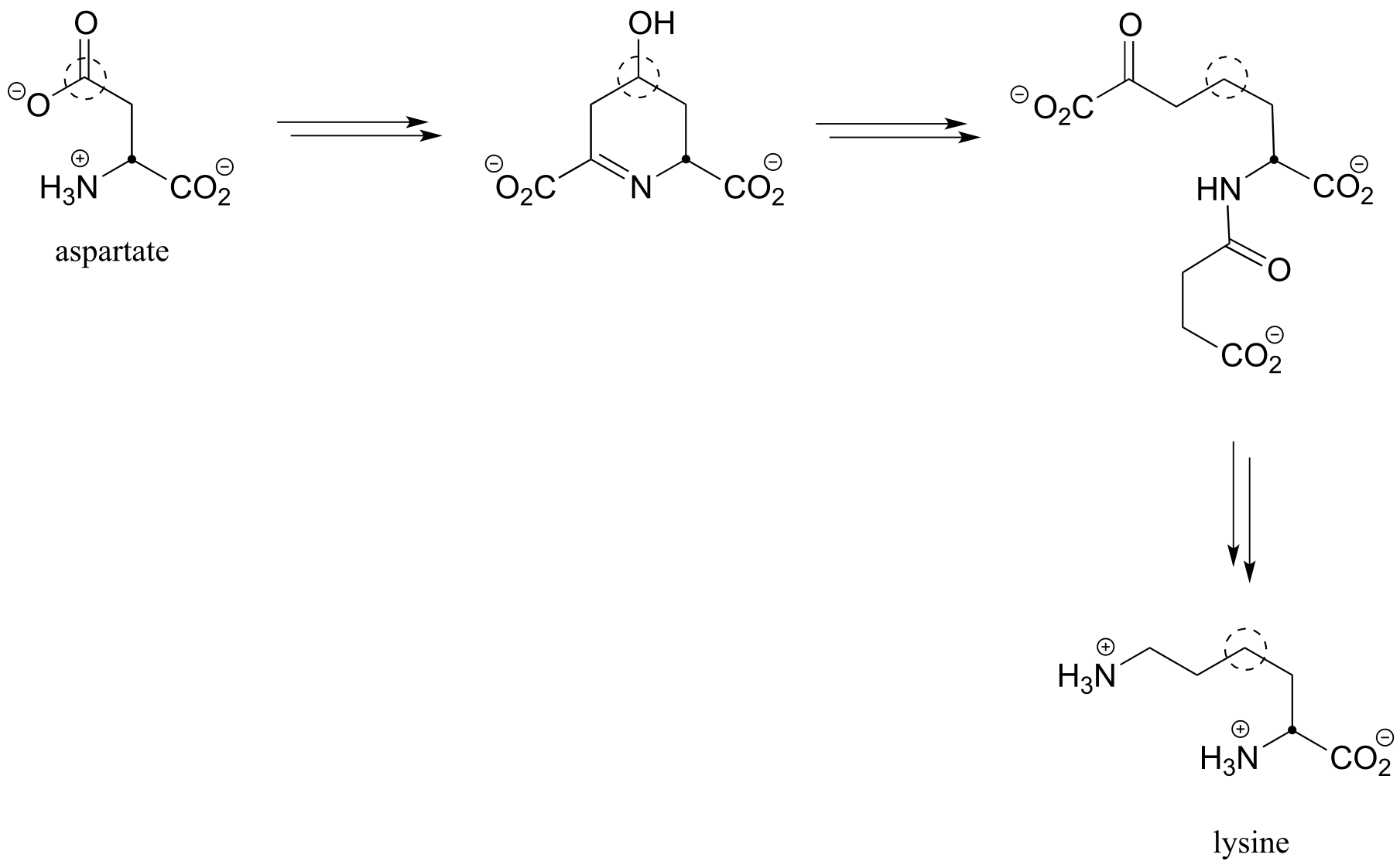
c) Below is an incomplete pathway diagram for the biosynthesis of the amino acid lysine, starting from aspartate. Fill in the missing steps and reactants/coenzymes to complete the diagram. The solid dot and dashed circle are provided to help you to trace two of the carbons from substrate to product.
d) Below is the second half of the tryptophan degradation pathway. Fill in the missing steps and reactants/coenzymes to complete the diagram.
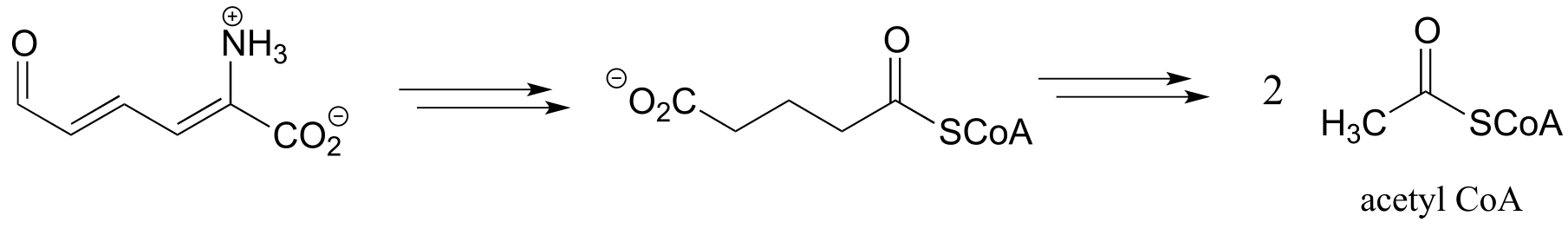
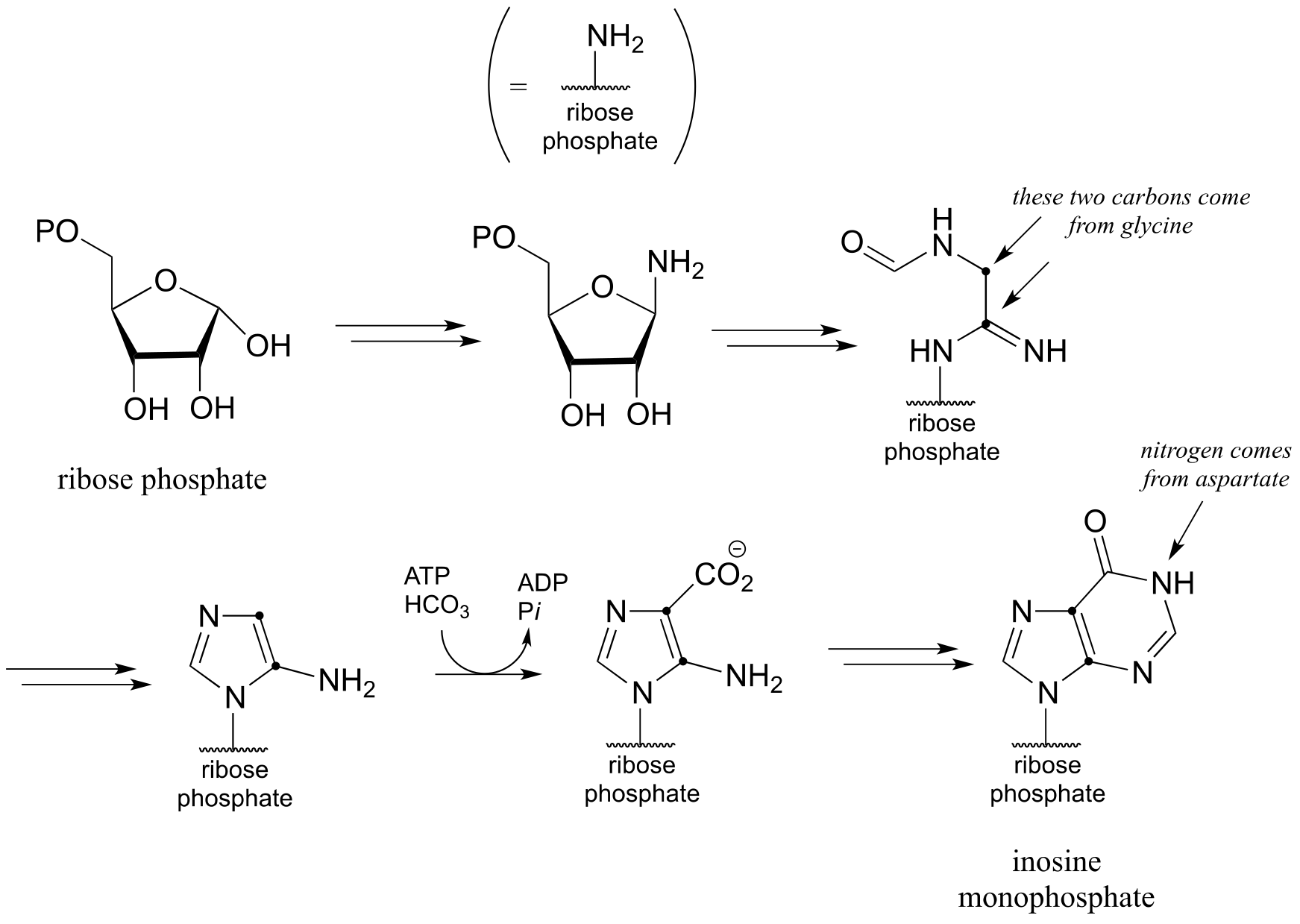
d) Below is an incomplete pathway diagram for the biosynthesis of inosine monophosphate, a precursor to the nucleotides adenosine monophosphate (AMP) and guanosine monophosphate (GMP). Fill in missing steps and reactants/coenzymes to complete the diagram. Note that one enzymatic step is provided (this is a carboxylation reaction of a type that we have not studied).
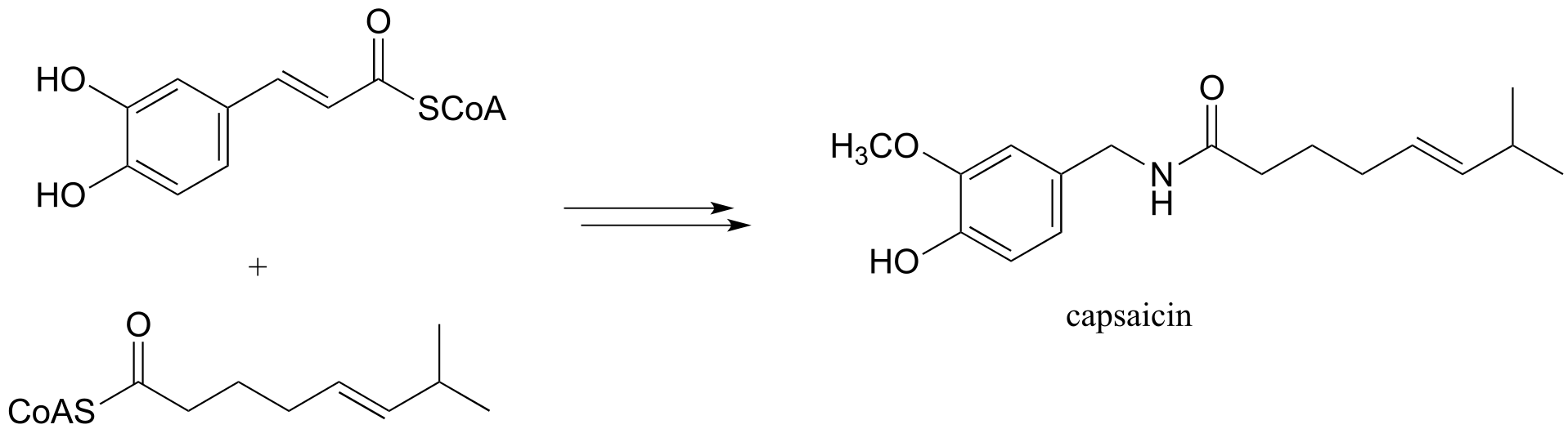
e) We begin our study of organic chemistry with a story about a hot pepper eating contest in Wisconsin (see the introduction the Chapter 1), and a compound called capsaicin which causes the ‘hot’ in hot peppers. As our last problem, let’s try to predict some of the key steps in the biosynthesis of capsaicin.
Phase 1:
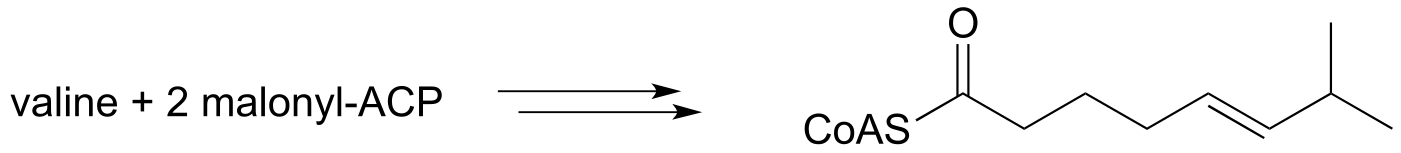
Phase 2: