14: Electrophilic reactions
Contents
14: Electrophilic reactions#
Memorial for Bridget Bishop, hanged as a witch in Salem, Massachusetts
photo credit: Melissa Nunez (https://www.flickr.com/photos/143702874@N05/)
Introduction#
Linnda Caporael probably should have paid a little more attention to the graduation requirements in her college catalog. Going through the graduation checklist during her senior year, she discovered to her dismay that she still needed to fulfill a social science requirement, so she promptly enrolled in an American History course. It was a decision that would in time lead to her authoring a paper in a prestigious scientific journal, being featured in a front page story in the New York Times, and changing our understanding of one of the most intriguing - and disturbing – episodes in American history.
Professor Caporael (Linnda went on to become a professor of Behavioral Psychology at Rensselaer Polytechnic Institute) recounted her story in an episode of the PBS documentary series Secrets of the Dead. Early in the semester, she learned that as part of her history course she would be required to complete a research paper on a topic of her own choosing. She had recently seen a performance of The Crucible, Arthur Miller’s classic play about the Salem witch trials, and decided to do her research on Anne Putnam, one of the young Salem girls who accused several village women of bewitching them. The symptoms of ‘bewitchment’ that afflicted the girls were truly frightening: thrashing and convulsions, visions of snakes and ferocious beasts, a sudden inability to speak, and a feeling that ants were crawling under their skin.
These children were bitten and pinched by invisible agents: their arms, necks and backs turned this way and that way, and returned back again, so as it was, impossible for them to do of themselves, and beyond the power of any epileptick fits, or natural disease to effect. Sometimes they were taken dumb, their mouth flopped, their throats choaked, their limbs wracked and tormented…
(From “A Modest Enquiry Into the Nature of Witchcraft” (1702) by Reverend John Hale. http://historyofmassachusetts.org/betty-parris-first-afflicted-girl-of-the-salem-witch-trials/)
As Linnda continued to read accounts of the ‘fits’ afflicting the Salem girls, she was suddenly struck by the similarities to another, more recent episode that she had read about. In the summer of 1951, in the French village of Pont Saint Esprit, a number of local people were simultaneously seized by hallucinations, convulsions, and other symptoms very much like those described during the Salem witch trials hundreds of years earlier. Leon Armunier, a former postman in Pont Saint Esprit, described his experience to the BBC:
It was terrible. I had the sensation of shrinking and shrinking, and the fire and the serpents coiling around my arms …Some of my friends tried to get out of the window. They were thrashing wildly. . . screaming, and the sound of the metal beds and the jumping up and down… the noise was terrible. I’d prefer to die rather than go through that again.
There have been several explanations offered for what caused the Pont Saint Esprit outbreak (including that the CIA was experimenting with mass LSD poisoning as a form of chemical warfare), but the most widely accepted theory is that the hallucinations were caused by eating bread made from contaminated grain.
Claviceps purpurea, a fungus known to grow in rye and other grains, produces a class of hallucinogenic compounds called ‘ergot alkaloids’ which are derived from lysergic acid (the hallucinogenic drug LSD is a synthetic derivative of lysergic acid). Claviceps thrives in damp grain, and special care must be taken to avoid contamination when storing grain grown during warm, rainy summers.
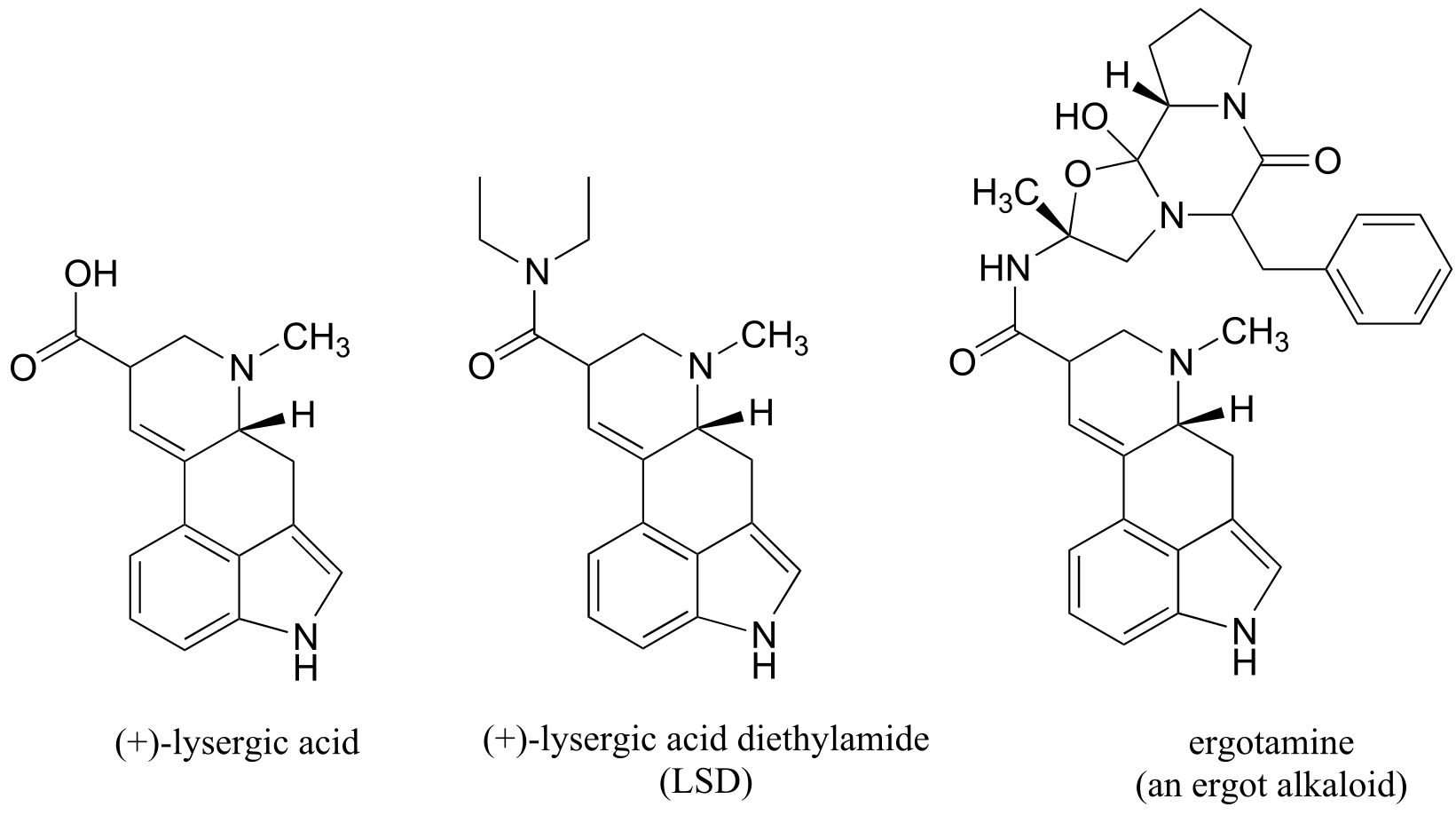
fig 1a
Digging deeper into the records of the Salem witchcraft episode, Linnda Caporael learned that the summer of 1691 was unusually damp. The first cases of ‘bewitchment’ in Salem village occurred in the winter of 1691-1692, when the villagers would have been consuming grain stored from the previous summer. Rye, the kind of grain most vulnerable to Claviceps contamination, was the staple crop in Salem at the time. Furthermore, nearly all of the affected girls lived on farms on the swampy western edge of the town, where Claviceps contamination would have been most likely to occur.
This was all circumstantial evidence, to be sure, but it was enough to convince Linnda that ergot poisoning was much more plausible as a root cause of the behavior of the afflicted girls than simply mass hysteria, which had long been the accepted theory. She summarized her findings in a paper that was later published in the journal Science, with the colorful title “Ergotism: The Satan Loosed in Salem?” (Science 1976, 192, 21). Her theory is still not universally accepted, but scientists and historians are for the most part in agreement that ergot poisoning was the cause of other outbreaks of convulsions and hallucinations, often called ‘Saint Anthony’s Fire’, that have occurred throughout European history. It is possible that ergot poisoning may have played a role in literature as well: professor Caporael, in an interview with PBS, recounts how she was recently contacted by an historian with in intriguing idea. Is it possible that Caliban, the wild, half-human character in Shakespeare’s The Tempest who is tormented by hallucinations inflicted upon him by the wizard Prospero, could be a literary manifestation of ergot poisoning episodes that occurred in England during Shakespeare’s time?
For every trifle are they set upon me;
Sometime like apes that mow and chatter at me
And after bite me, then like hedgehogs which
Lie tumbling in my barefoot way and mount
Their pricks at my footfall; sometime am I
All wound with adders who with cloven tongues
Do hiss me into madness.
*(*William Shakespeare’s The Tempest, Act II scene ii)
In this chapter, we will learn about a class of organic reaction that is central to the biosynthesis of ergot alkaloids in Claviceps. The key first step in the biosynthetic pathway is a reaction unlike any we have yet seen:
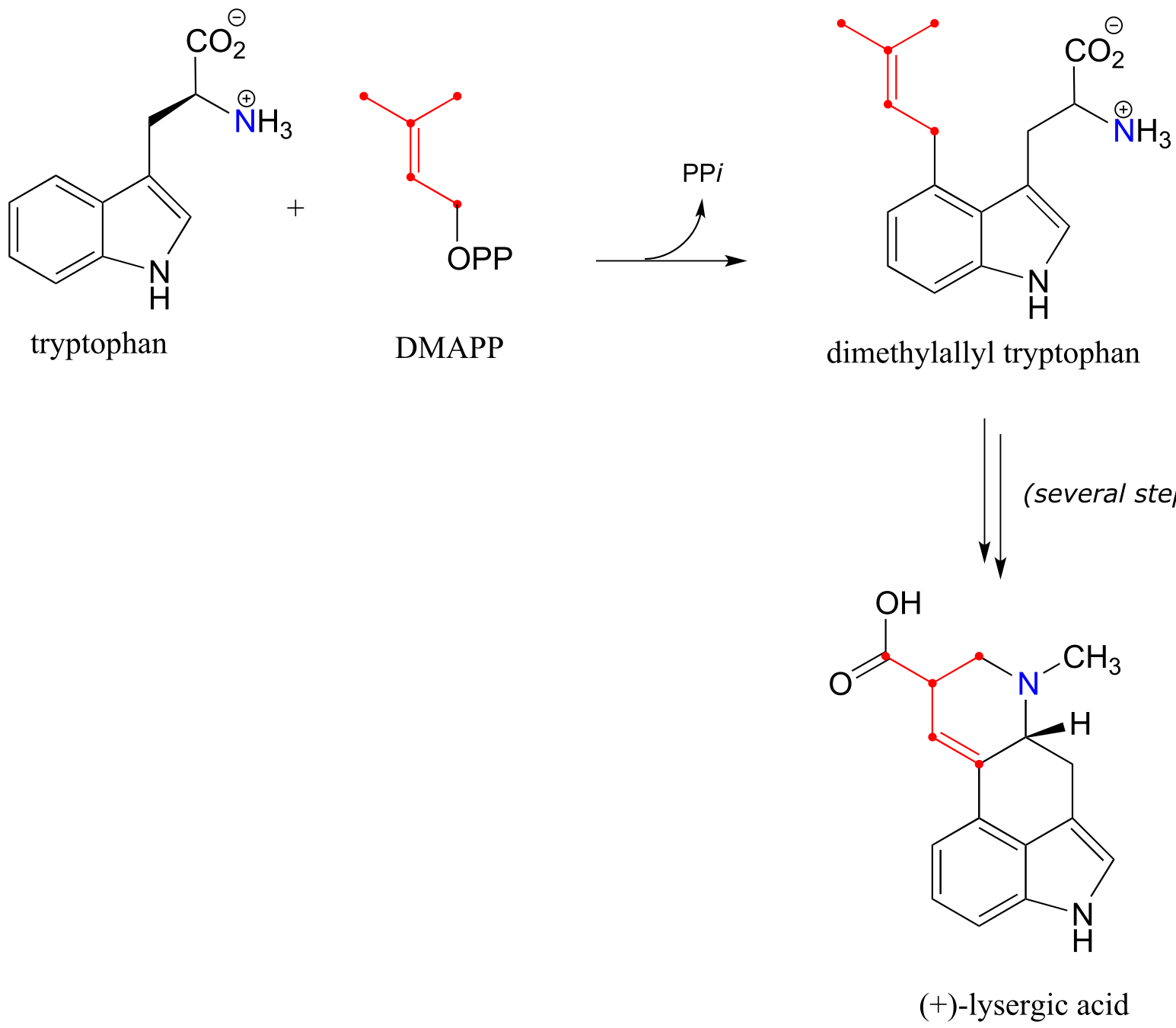
fig 1b
As you can see, the first step is condensation between the amino acid tryptophan and dimethylallyl diphosphate (DMAPP), the building block molecule for isoprenoids (section 1.3A). What you should also notice in the reaction figure above is that a new carbon-carbon bond is formed, and yet the chemistry involved is clearly very different from the carbon-carbon bonding forming aldol additions and Claisen condensations we learned about in the previous two chapters: there is no carbonyl to be found anywhere near the site of reaction, and one of the bond-forming carbons is part of an aromatic ring.
We will see later in this chapter that this reaction mechanism is classified as an ‘electrophilic aromatic substitution’, and is one of a broader family of organic reaction mechanisms that includes electrophilic additions, substitutions, and isomerizations. ‘Electrophilic’ is the key term here: in organic chemistry, an ‘electrophilic’ reaction mechanism is one in which the π-bonded electrons in a carbon-carbon double (or sometimes triple) bond are drawn towards an electron-poor species, often an acidic proton or carbocation. In essence, the π bond is acting as a nucleophile or base.
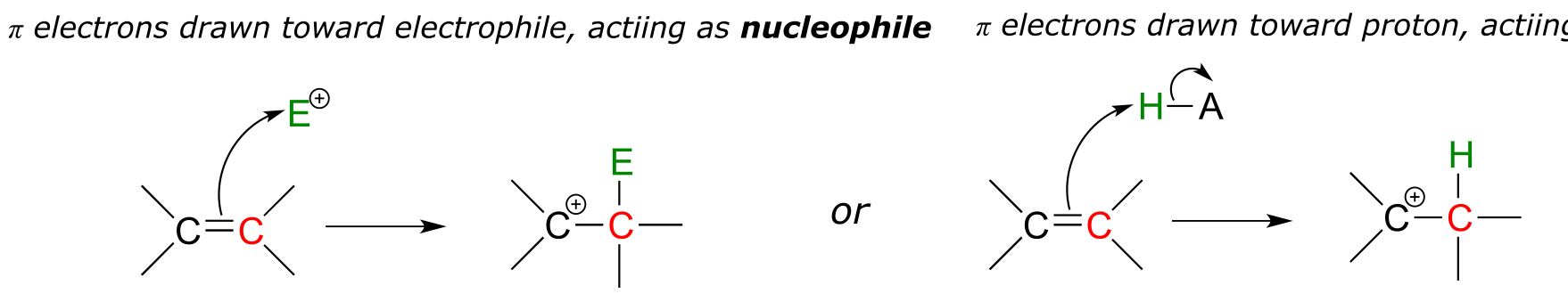
fig 1
Notice above that, once the π bond breaks and a new bond forms, the second carbon that was part of the original π bond becomes a carbocation. Carbocation intermediates play a critical role in this chapter, because a carbocation is a highly reactive species and will quickly attract a pair of electrons. The stability of the carbocation intermediate (recall that we learned about carbocation stability in section 8.5), and the manner in which it accepts a pair of electrons, plays a key role in determining the outcome of the reaction.
Section 14.1: Electrophilic addition#
14.1A: Addition of HBr to alkenes#
The simplest type of electrophilic reaction to visualize is the addition of a haloacid such as HBr to an isolated alkene. It is not a biological reaction, but nonetheless can serve as a convenient model to introduce some of the most important ideas about electrophilic reactions.
Electrophilic addition of HBr to an alkene:
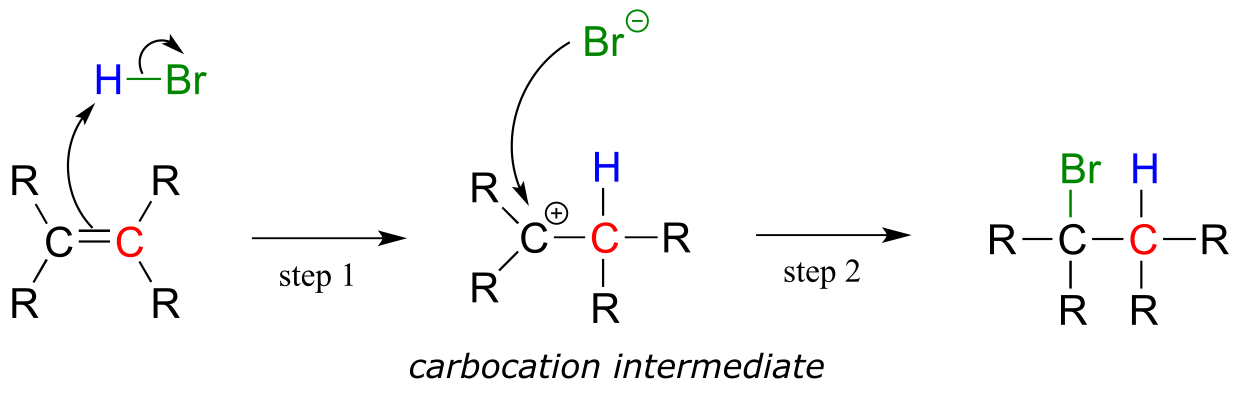
fig 1
Step 1 is an acid-base reaction: the π electrons of the alkene act as a base and extract the acidic proton of HBr. This leaves one of the carbons with a new bond to hydrogen, and the other with an incomplete octet and a positive formal charge. In step 2, the nucleophilic bromide anion attacks the electrophilic carbocation to form a new carbon-bromine bond. Overall, the HBr molecule - in the form of a proton and a bromide anion - has been added to the double bond.
To understand how π-bonded electrons in an alkene could be basic, let’s first review the bonding picture for alkenes. Recall (section 2.1C) that the both of the carbons in an alkene group are sp2 hybridized, meaning that each carbon has three sp2 hybrid orbitals extending out in the same plane at 180o angles (trigonal planar geometry), and a single, unhybridized p orbital oriented perpendicular to that plane - one lobe above the plane, one lobe below.
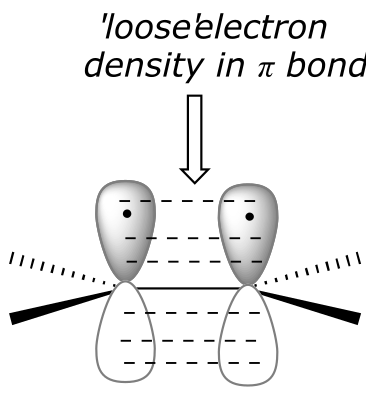
fig 2
The unhybridized p orbitals on the two alkene carbons overlap, in a side-by-side fashion, to form the π bond, which extends above and below the plane formed by the σ bonds. The two electrons shared in this π bond are farther away from the carbon nuclei than the electrons in the carbon-carbon σ bond, and thus are more accessible to the acidic proton. In addition, recall that molecular orbital (MO) theory tells us that π orbitals are higher in energy than σ orbitals (section 2.2). As a consequence, it is easier to break the π bond of an alkene than it is to break the σ bond: the π bond is more reactive.
As the HBr molecule approaches the alkene, a new σ bond is formed between one of the alkene carbons and the electron-poor proton from HBr. The carbon, which was sp2 hybridized when it was part of the alkene, is now sp3 hybridized. The other alkene carbon is still sp2 hybridized, but it now bears a positive formal charge because it has only three bonds, and its p orbital is empty. But it won’t stay empty for long: a carbocation is a very reactive, unstable intermediate. The bromide ion will rapidly act as a nucleophile, filling the orbital with a pair of electrons, and now with four σ bonds the carbon is sp3-hybridized.
The first step in the electrophilic addition reaction is much slower than the second step, because the intermediate carbocation species is higher in energy than either the reactants or the products, and as a result the energy barrier for the first step is also higher than for the second step. The slower first step is the rate-determining step: a change in the rate of the slow step will effect the rate of the overall reaction, while a change in the rate of the fast step will not.
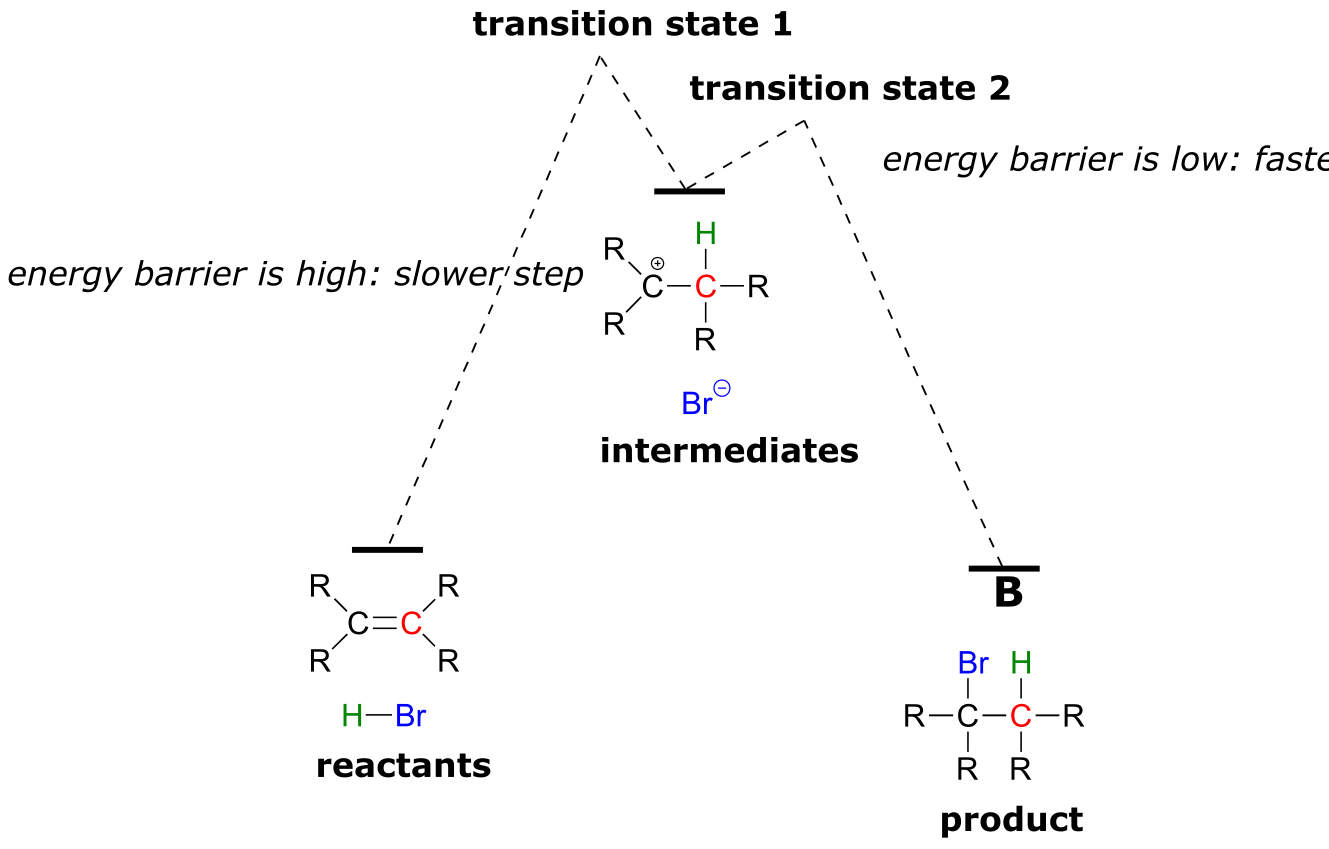 (fig 3)
(fig 3)
fig 2a
It is important to recognize the inherent difference between an electrophilic addition to an alkene and a conjugate addition to an alkene in the position, the latter of which we studied earlier in section 13.4. In both reactions, a proton and a nucleophile add to the double bond of an alkene. In a conjugate addition, the nucleophilic attack takes place first, resulting in a negatively charged intermediate (an enolate). Protonation is the second step. Also, of course, the alkene must be conjugated to a carbonyl or imine.
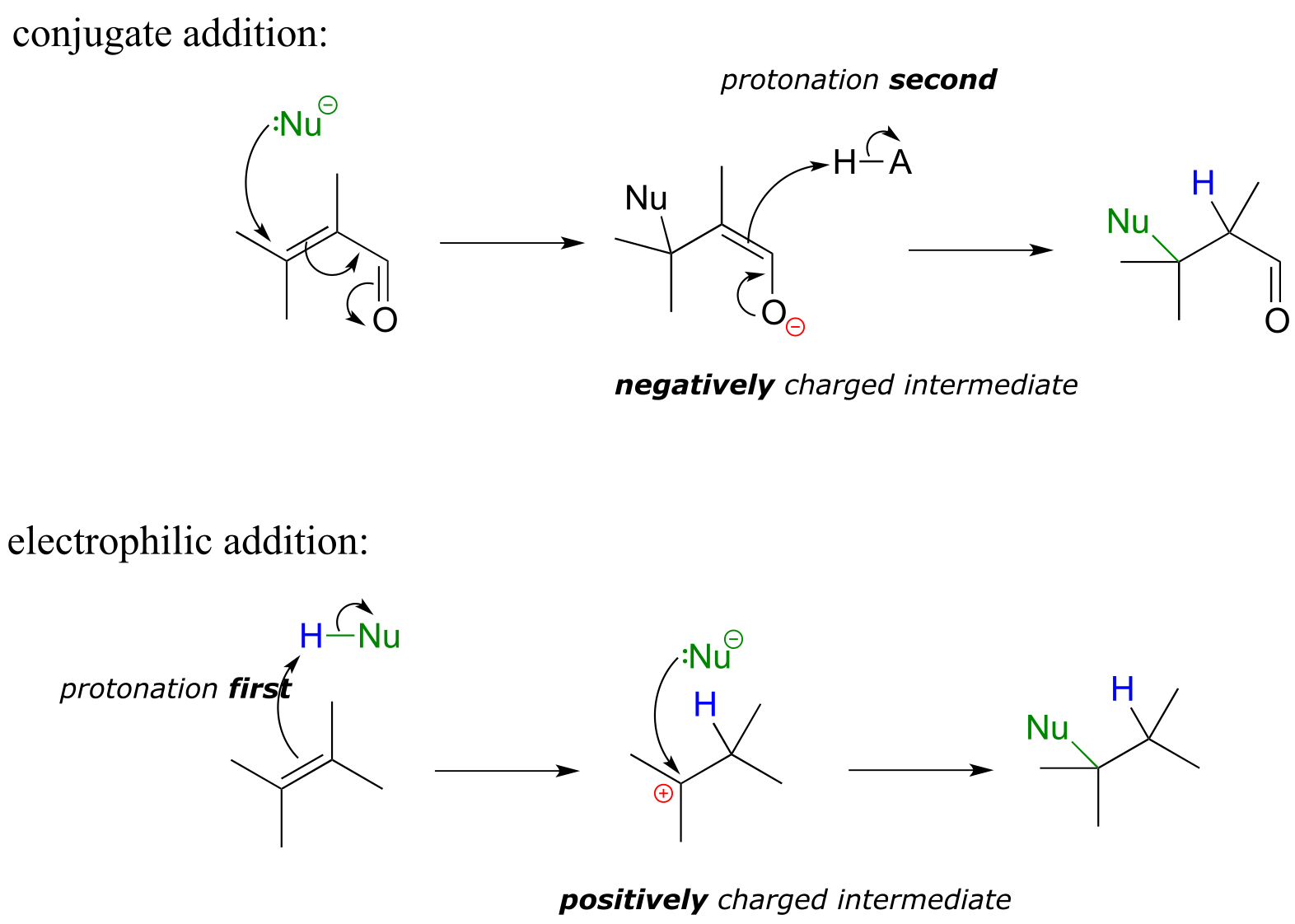
fig 3
In an electrophilic addition, proton abstraction occurs first, generating a positively-charged intermediate. Nucleophilic attack is the second step. No conjugated carbonyl or imine group is required: in fact a nearby carbonyl group would actually slow down a hypothetical electrophilic addition reaction down because a carbonyl is an electron withdrawing, carbocation-destabilized group.
14.1B: The stereochemistry of electrophilic addition#
Depending on the structure of the starting alkene, electrophilic addition has the potential to create two new chiral centers. Addition of HBr to an alkene is not stereoselective: the reaction results in racemization at both of the alkene carbons. Consider the addition of HBr to cis-3,4-dimethyl-3-hexene. The initial proton abstraction step creates a new chiral center, and because the acidic proton could be added to either side of the planar alkene carbon with equal probability, the center could have either S or R configuration.
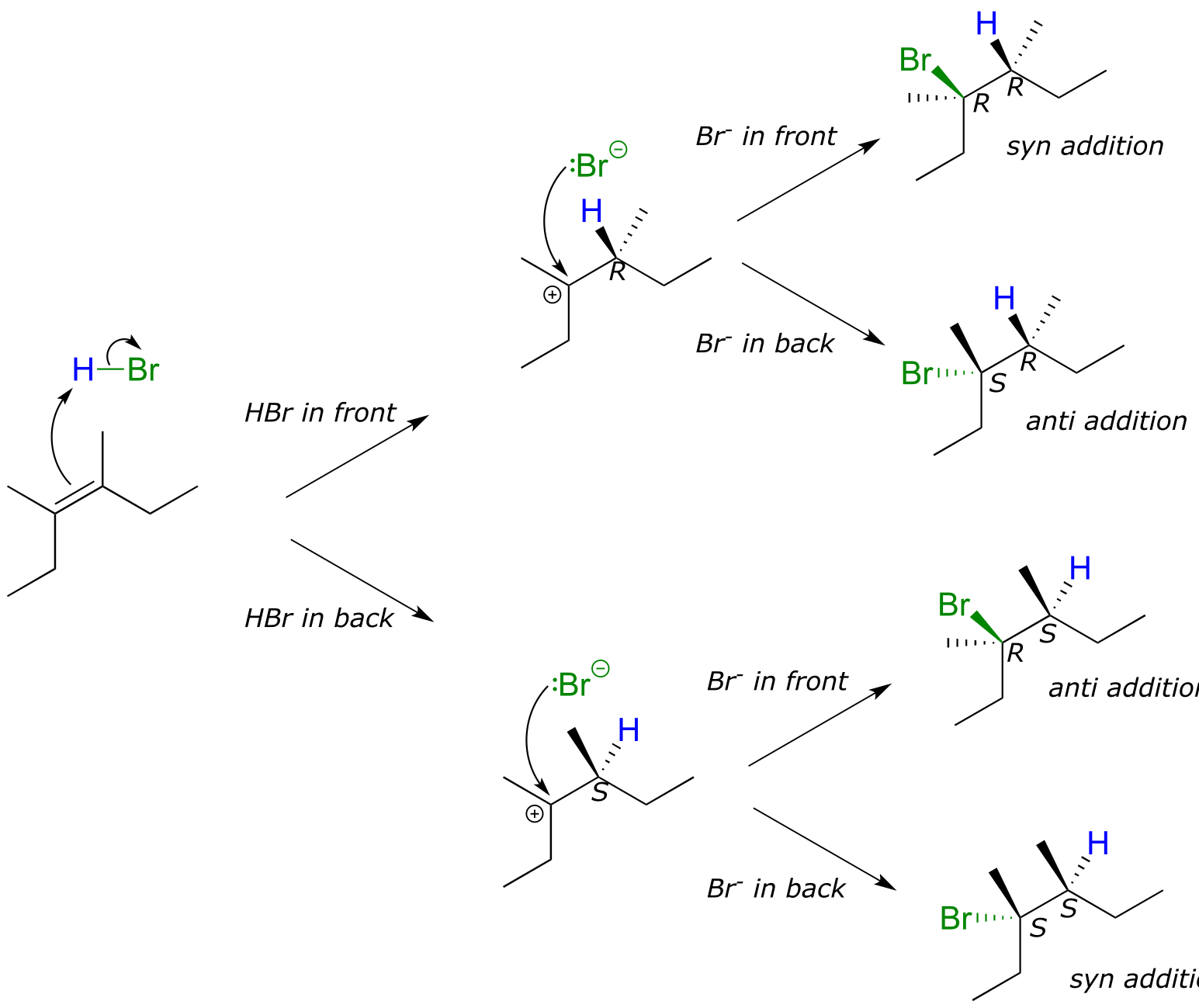
fig 4
Likewise, in the second step the nucleophilic bromide ion could attack from either side of the planar carbocation, leading to an equal mixture of S and R configuration at that carbon as well. Therefore, we expect the product mixture to consist of equal amounts of four different stereoisomers.
Exercise 14.1: Predict the product(s) of electrophilic addition of HBr to the following alkenes. Draw all possible stereoisomers that could form, and take care not to draw identical structures twice.
a) trans-2-butene b) cis-3-hexene c) cyclopentene
14.1C: The regiochemistry of electrophilic addition#
In many cases of electrophilic addition to an alkene, regiochemistry comes into play: the reaction can result in the formation of two different constitutional isomers. Consider the electrophilic addition of HBr to 2-methylpropene:
(fig 4)
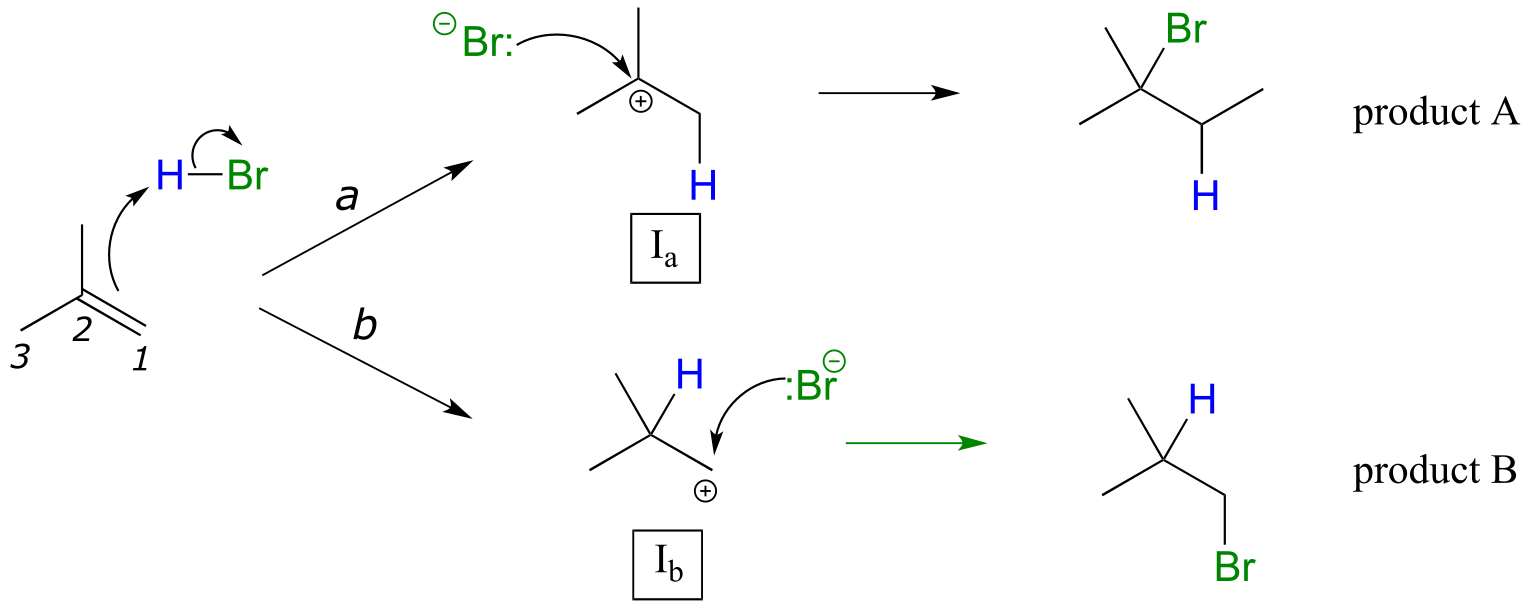
fig 5
Note that carbon #1 and carbon #2 in the starting alkene are not the same - carbon #2 is bonded to two methyl groups, and carbon #1 to two hydrogen atoms. The initial protonation step could therefore go two different ways, resulting in two different carbocation intermediates. Notice how pathway ‘a’ gives a tertiary carbocation intermediate (Ia), while pathway ‘b’ gives a primary carbocation intermediate (Ib) We know from section 8.5 that the tertiary carbocation Ia is lower in energy. Consequently, the transition state TS(a) leading to Ia is lower in energy than TS(b), meaning that Ia forms faster than Ib.
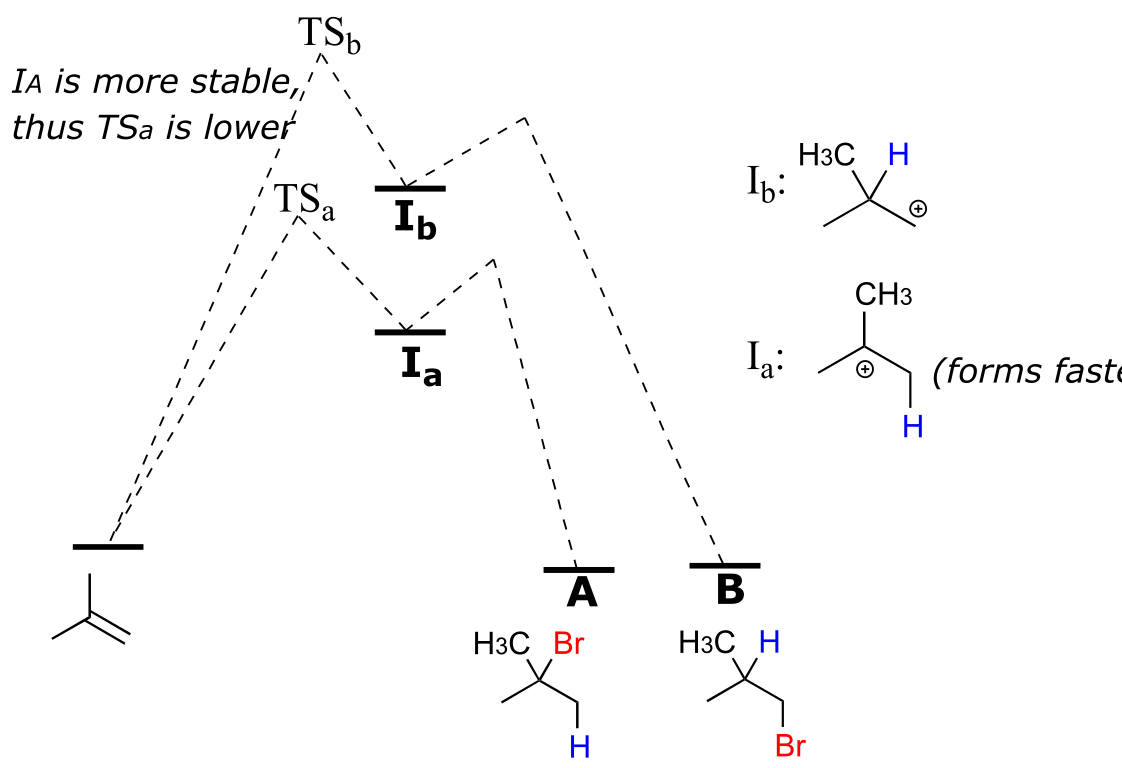
fig 6
Because the protonation step is the rate determining step for the reaction, tertiary alkyl bromide A will form faster than the primary alkyl bromide B, and thus A will be the predominant product of the reaction. The electrophilic addition of HBr to 2-methylpropene is regioselective: more than one constitutional isomer can potentially form, but one isomer is favored over the other. It is generally observed that in electrophilic addition of haloacids to alkenes, the more substituted carbon is the one that ends up bonded to the heteroatom of the acid, while the less substituted carbon is protonated. This ‘rule of thumb’ is known as Markovnikov’s rule, after the Russian chemist Vladimir Markovnikov who proposed it in 1869.
While it is useful in many cases, Markovikov’s rule does not apply to all electrophilic addition reactions. It is better to use a more general principle:
The regioselectivity of electrophilic addition
When an asymmetrical alkene undergoes electrophilic addition, the product that predominates is the one that results from the more stable of the two possible carbocation intermediates.
How is this different from Markovnikov’s original rule? Consider the following hypothetical reaction, in which the starting alkene incorporates two trifluoromethyl substituents:
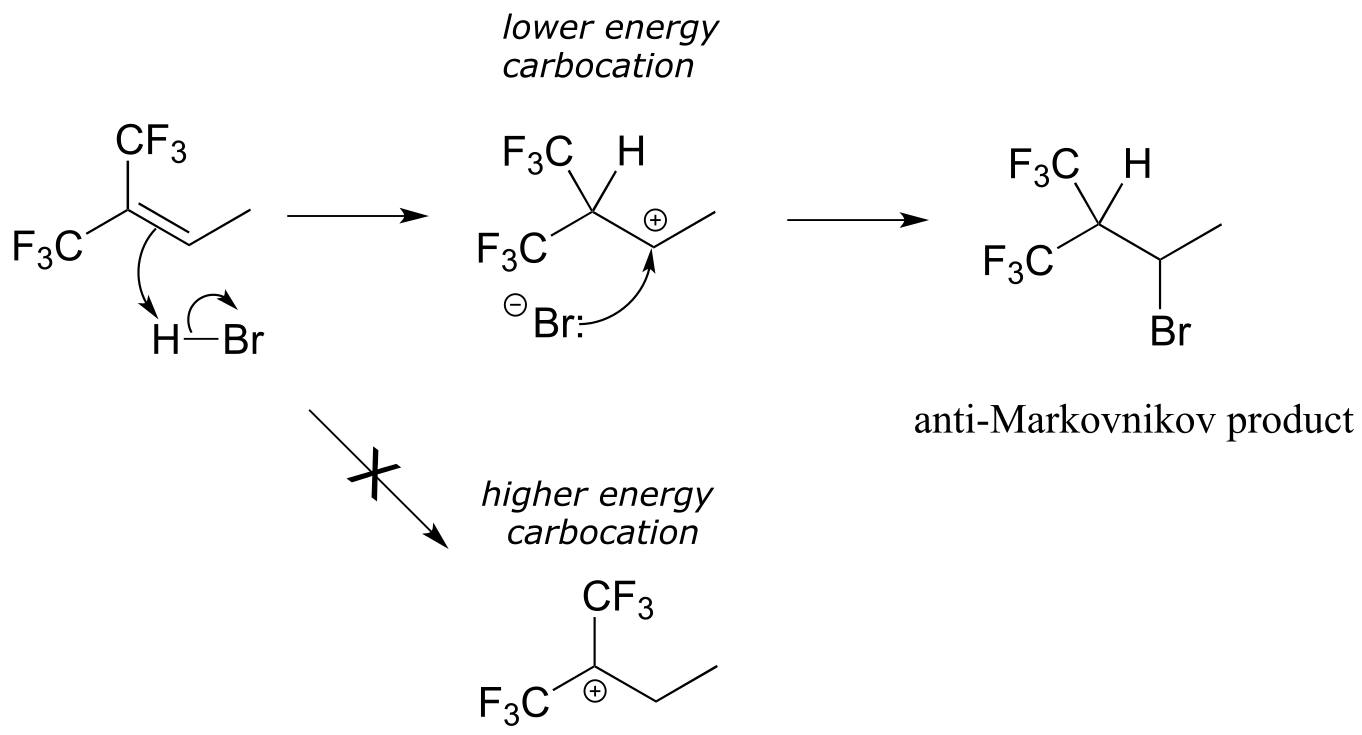
fig 7
Now when HBr is added, it is the less substituted carbocation that forms faster in the rate-determining protonation step, because in this intermediate the carbon bearing the positive charge is located further away from the electron-withdrawing, cation-destabilizing fluorines. As a result, the predominant product is the secondary rather than the tertiary bromoalkane. This is referred to as an anti-Markovnikov addition product, because it ‘breaks’ Markovnikov’s rule.
If the two possible carbocation intermediates in an electrophilic addition reaction are of similar stability, the product will be a mixture of constitutional isomers.
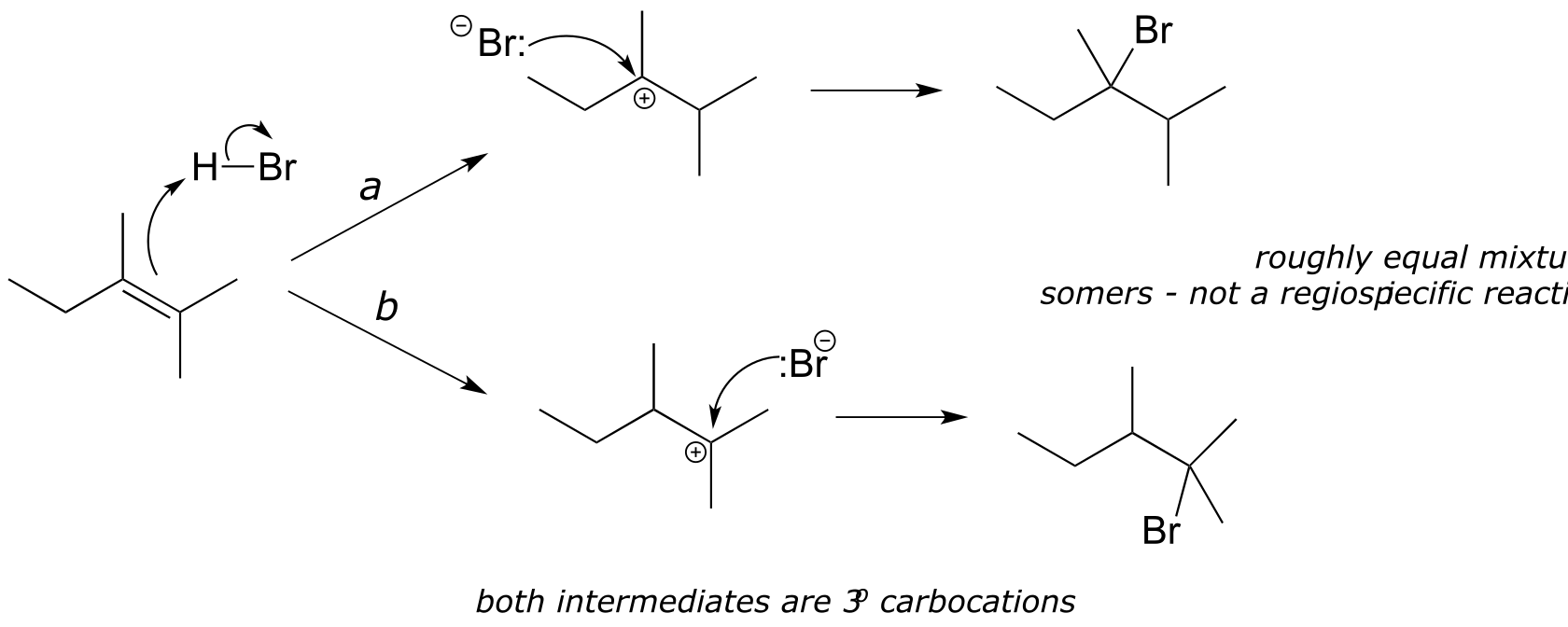
fig 8
14.1D: Electrophilic addition of water and alcohol#
The (non-biochemical) addition of water to an alkene is very similar mechanistically to the addition of a haloacid such as HBr or HCl, and the same stereochemical and regiochemical principles apply. A catalytic amount of a strong acid such as phosphoric or sulfuric acid is required, so that the acidic species in solution is actually H3O+. Note that H3O+ is regenerated in the course of the reaction.
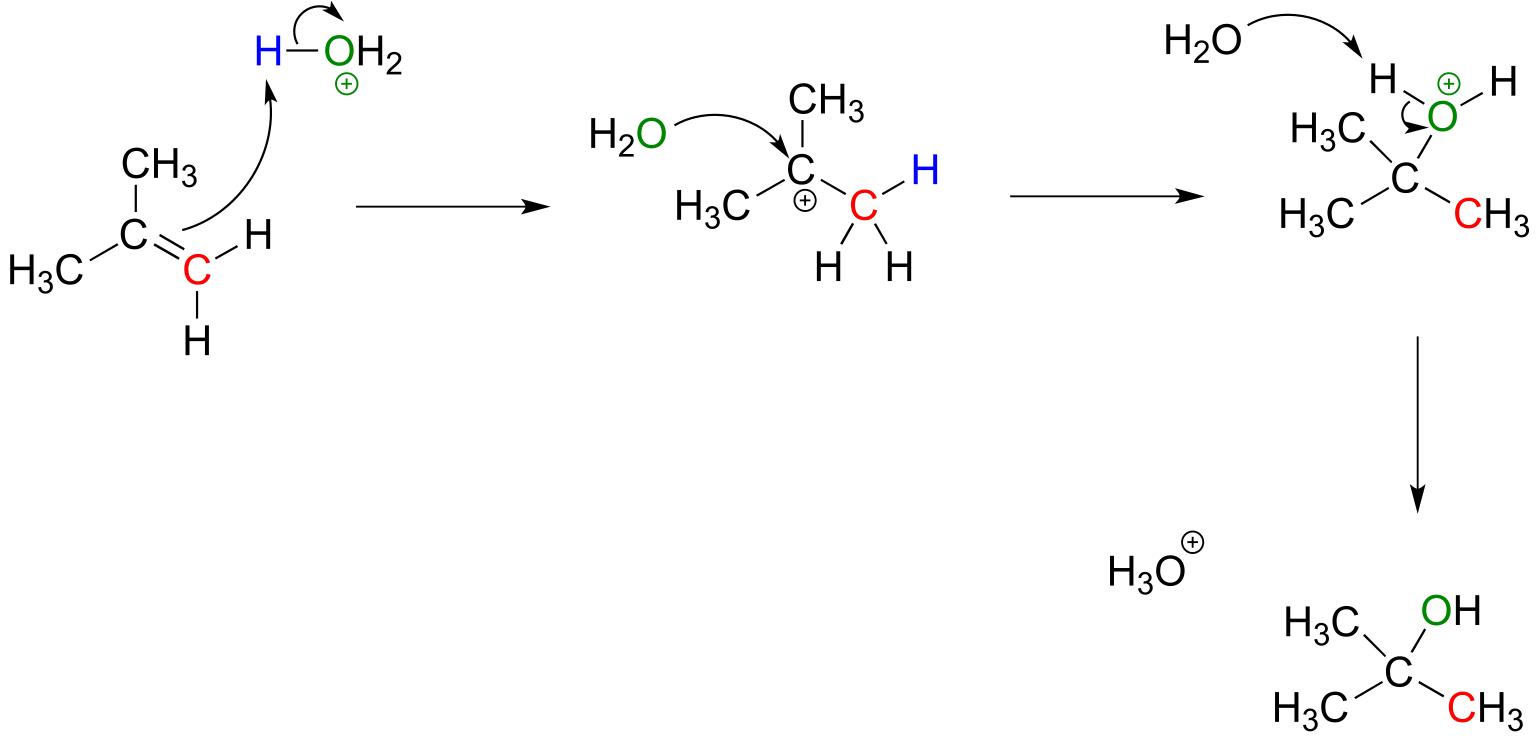
fig 9
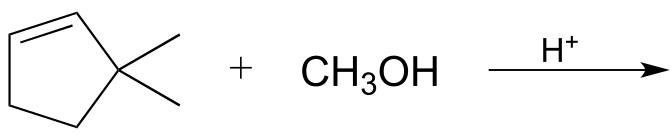
If an alkene is treated with methanol and a catalytic amount of strong acid, the result is an ether:

fig 10
Exercise 14.2: Draw a mechanism for the ether-forming reaction above.
14.1E: Addition to conjugated alkenes#
Electrophilic addition to conjugated alkenes presents additional regiochemical possibilities, due to resonance delocalization of the allylic carbocation intermediate. Addition of one molar equivalent of HBr to 1,3-butadiene, for example, leads to a mixture of three products, two of which are a pair of enantiomers due to the creation of a chiral center at carbon #2.
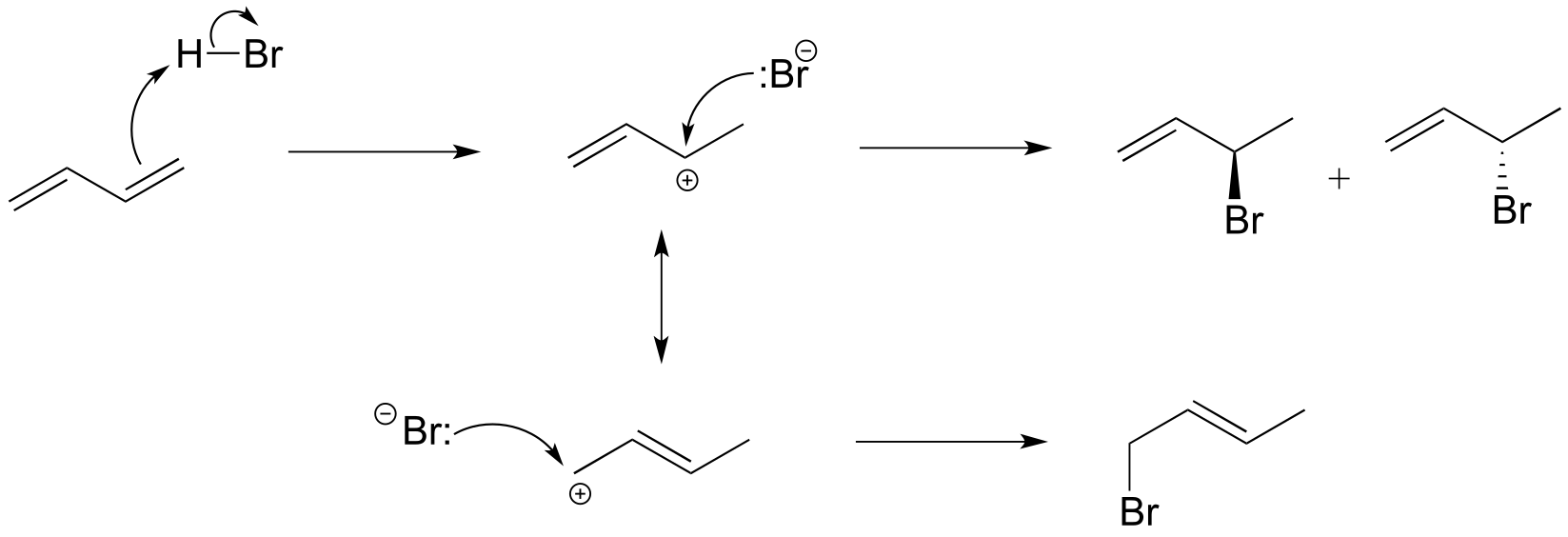
Exercise 14.3: Explain why 4-bromo-1-butene is not a significant product of the reaction above.
Exercise 14.4: Predict the major product(s) of the following reactions. Draw all possible stereoisomers, and take care not to draw the same structure twice.
a)

b)

c)
d) Hint - are the double bonds in an aromatic ring likely to undergo electrophilic addition?
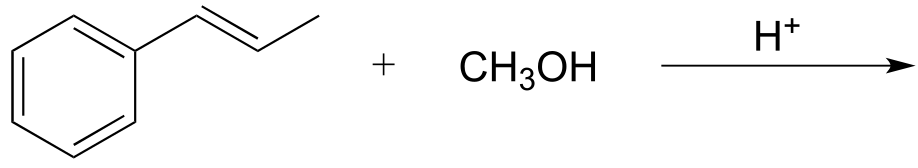
e)
ig 12
14.1F: Biochemical electrophilic addition reactions#
Myrcene is an isoprenoid compound synthesized by many different kinds of plants and used in the preparation of perfumes. Recently an enzymatic pathway for the degradation of myrcene has been identified in bacteria (J. Biol. Chem 2010, 285, 30436). The first step of this pathway is electrophilic addition of water to a conjugated alkene system.
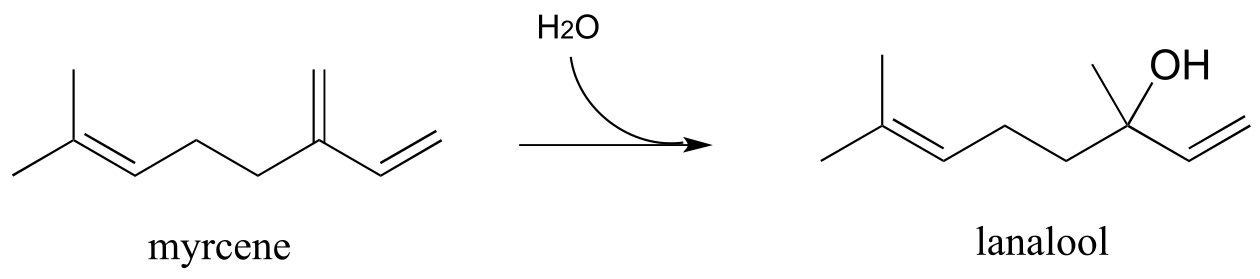
fig 15
Exercise 14.5: Draw a mechanism for the above reaction, showing two resonance contributors of the carbocation intermediate. How would you characterize the intermediate?
Although the hydration of myrcene above looks very familiar, many enzyme-catalyzed electrophilic addition reactions differ from what we have seen so far, in that the electron-poor species attacked by the π-bonded electrons in the initial step is a carbocation rather than an acidic proton:
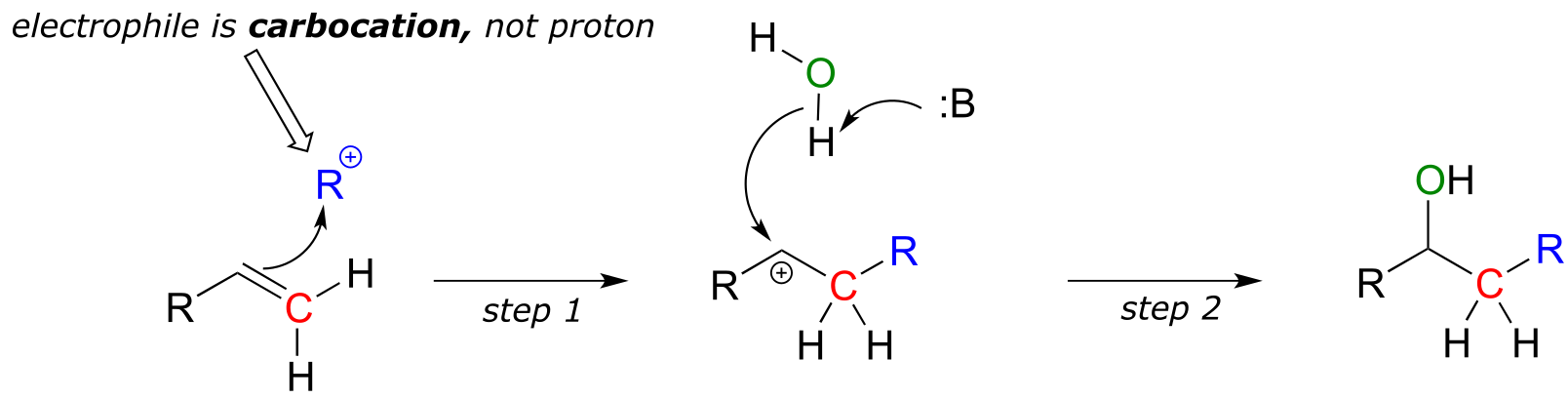
fig 13
Alpha-terpineol, a major component in the sap of pine trees, is formed in an electrophilic addition reaction. The first thing that happens (which we will refer to below as ‘step a’, in order to keep the step numbering consistent what the addition mechanisms we have seen so far) is departure of a pyrophosphate leaving group, forming an allylic carbocation electrophile.
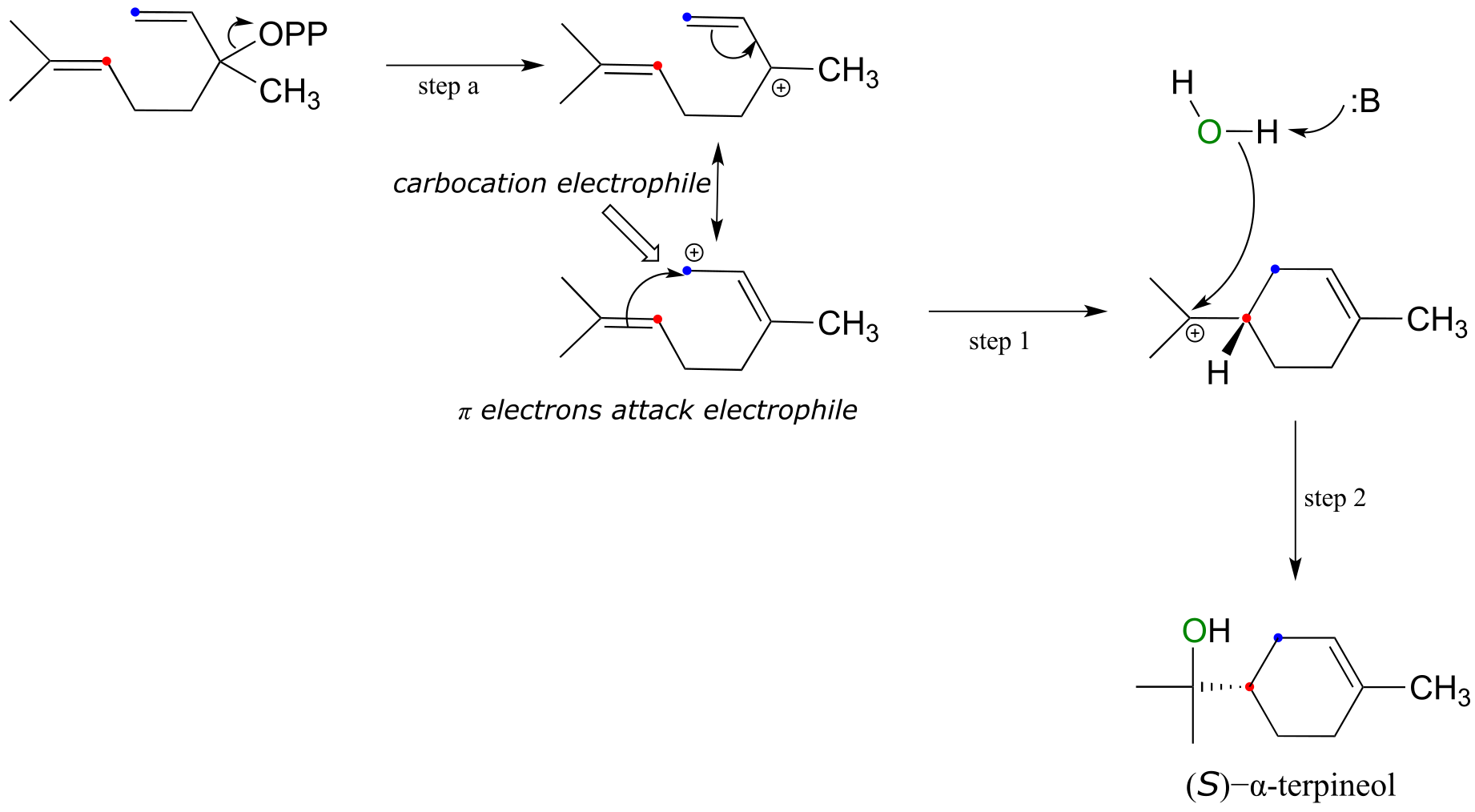
fig 14
The actual electrophilic addition stage of the reaction begins with step 1, as the π electrons an alkene are drawn toward one of the two carbons that share the positive charge, effectively closing a six-membered ring. A water molecule then attacks the second carbocation intermediate (step 2), which completes the addition process.
Notice something important about the regiochemical course of the reaction: step 1 results in the formation of a six-membered ring and a tertiary carbocation. As we have stressed before, biochemical reactions tend to follow energetically favorable mechanistic pathways.
Exercise 14.6: An alternate regiochemical course to step 1 shown above could result in a seven-membered ring and a secondary carbocation, a much less energetically favorable intermediate in terms of both carbocation stability and ring size. Draw a mechanism for this hypothetical alternate reaction, and show the product that would result after the addition of water in a hypothetical ‘step 2’.
Section 14.2: Elimination by the E1 mechanism#
14.2A: E1 elimination - an overview#
The reverse of electrophilic addition is called E1 elimination. We will begin by looking at some non-biochemical E1 reactions, as the E1 mechanisms is actually somewhat unusual in biochemical pathways.
E1 elimination:
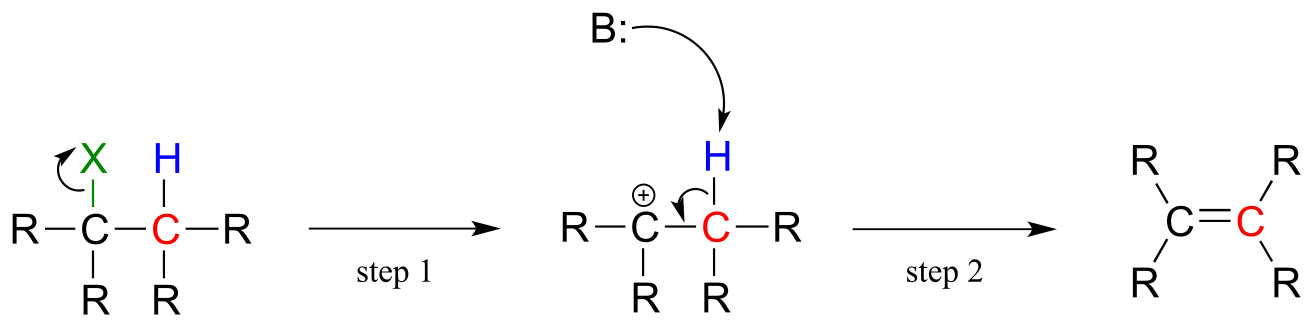
fig 16
An E1 elimination begins with the departure of a leaving group (designated ‘X’ in the general figure above) and formation of a carbocation intermediate (step 1). Abstraction of a proton from an adjacent carbon (step 2) sends two electrons down to fill the empty p orbital of the carbocation, forming a new π bond. The base in this step may be the leaving group, or another basic species in solution.
E1 elimination does not occur when the leaving group is bonded to a primary carbon, unless the carbon is in the allylic or benzylic position. Recall that a primary carbocation, unless stabilized by resonance, is highly unstable and an unlikely reaction intermediate.
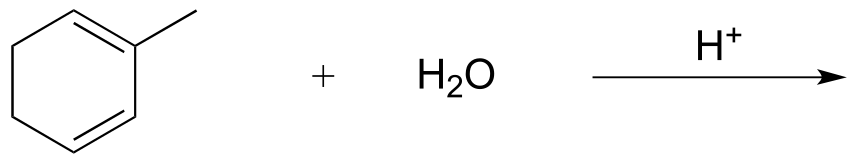
E1 eliminations can occur at secondary carbons, however. If cyclohexanol is heated with a catalytic amount of phosphoric acid, elimination of water (dehydration) results in cyclohexene as the product. The role of the phosphoric acid is to protonate the alcohol (‘step a’ below), making it a viable leaving group.

fig 17
The reaction is reversible, but if cyclohexene is distilled away from the reaction mixture as it forms, the equilibrium can be driven towards product (you may want to review Le Chatelier’s principle in your General Chemistry textbook). Separation of cyclohexene (boiling point 83 oC) from cyclohexanol (boiling point 161 oC) is simple because of the large difference in boiling points between the two liquids.
Exercise 14.7: When the laboratory reaction described above is run to completion, a viscous ‘goop’ is usually left over in the distillation flask, which hardens upon cooling. Draw a mechanism showing how this ‘goop’ might form, and explain why it hardens upon cooling.
14.2B: Regiochemistry of E1 elimination#
Nonenzymatic E1 reactions can often result in a mixture of more than one alkene product. Elimination of ‘HX’ from the following starting compound, for example, could yield three different possible alkene products.
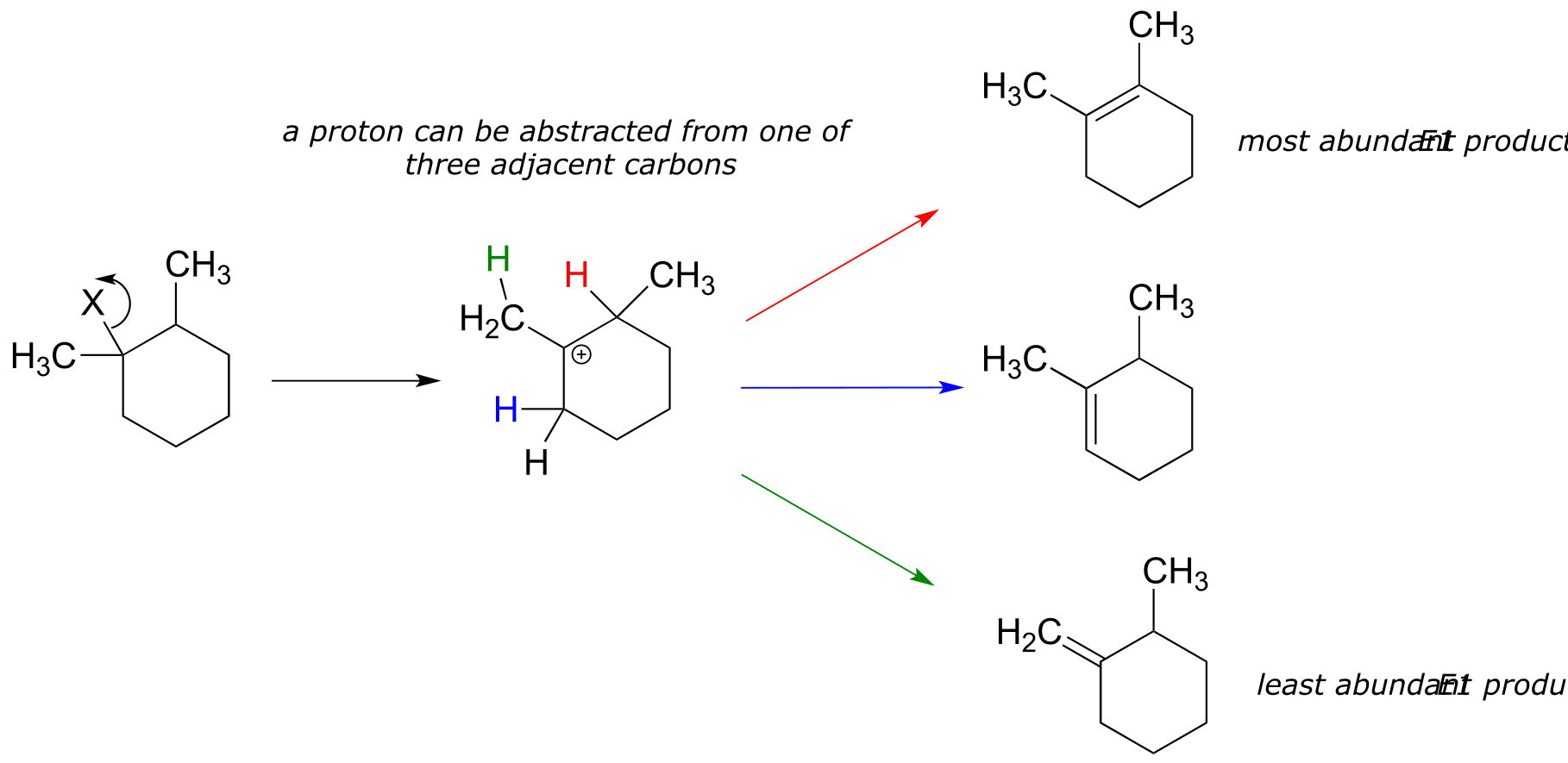
fig 18
Notice in the figure above that the three possible E1 products do not form in equal abundance. The most abundant alkene product is that which is most substituted: in other words, the alkene in which the two sp2 carbons are bonded to the fewest hydrogen atoms. In this case, the most substituted alkene has zero hydrogen substituents. The least substituted - and least abundant - alkene product has two hydrogen substituents, while the middle alkene has one hydrogen substituent. This trend is observed generally with nonenzymatic E1 elimination reactions, and is known as Zaitsev’s rule after the Russian chemist Alexander Zaitsev.
14.2C: Stereochemistry of E1 elimination#
Nonenzymatic E1 reactions can also result in both cis and trans alkenes. Keeping in mind that in general trans alkenes are more stable than cis alkenes, we can predict that trans alkenes will predominate in the product mixture.
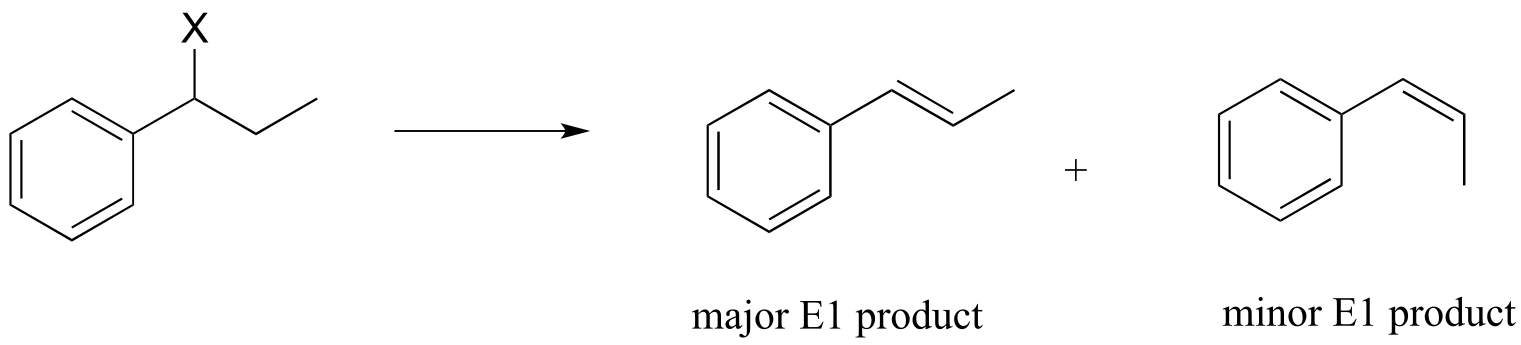
fig 19
Exercise 14.8: Draw the structures of all possible E1 products starting with the compounds below, and rank them in order of highest to lowest abundance.
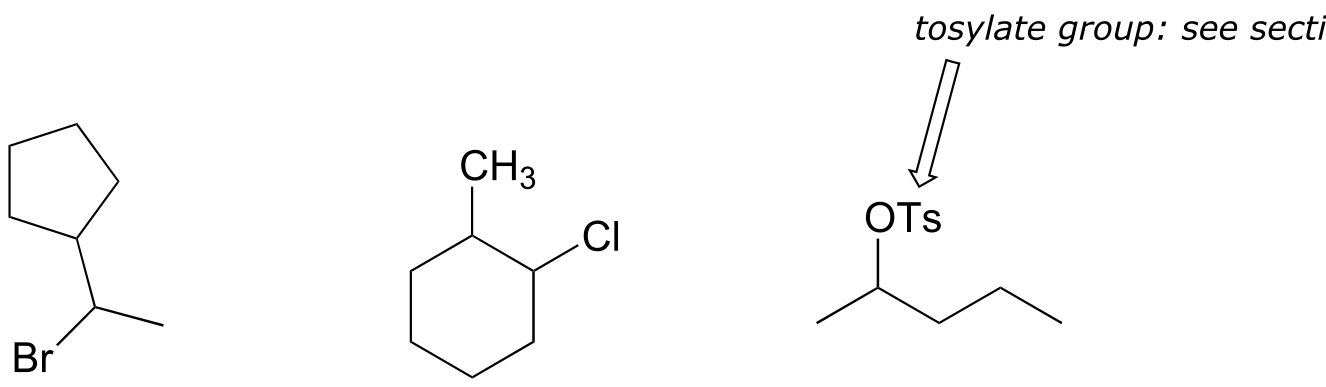
fig 20
14.2D: The E2 elimination mechanism#
When a strong base is combined with an alkyl halide (or alkyl tosylate/mesylate), elimination generally occurs by the E2 pathway, in which proton abstraction and loss of the leaving group occur simultaneously, without an intervening carbocation intermediate:
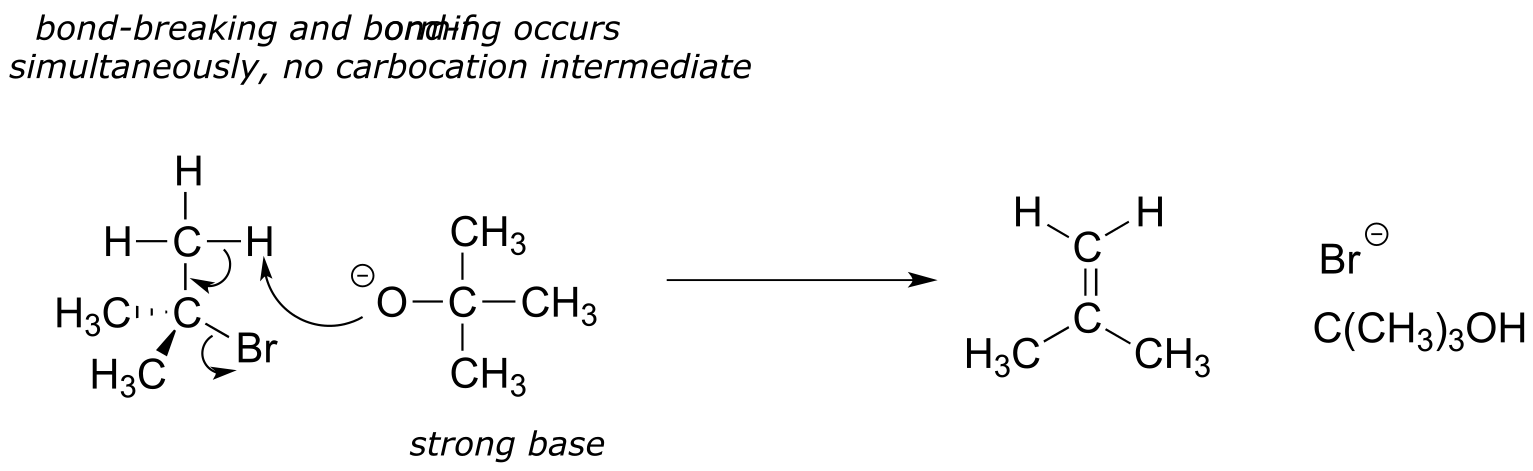
fig 20a
Just like in the SN2 mechanism, the ‘2’ in the E2 designation refers to the idea that the rate-determining (and only) step of the reaction is a collision between the two reacting molecules, in this case bromocyclohexane and methoxide ion.
14.2E: Competition between elimination and substitution#
Consider a reaction between water and bromocyclohexane. Based on what we have just learned, a likely product would be the alkene formed from an E1 elimination reaction (pathway (a) in red below).
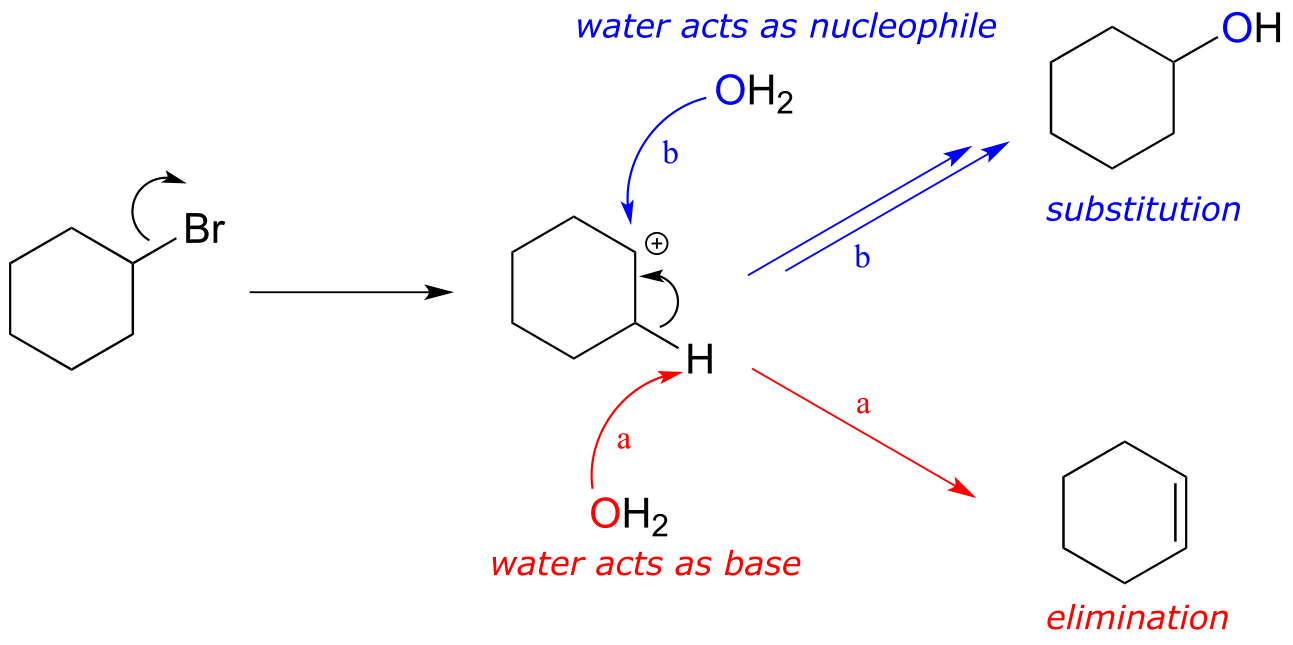
fig 20c
However, the reaction could take another course: what if the water molecule, instead of acting as a base, were to act as a nucleophile (pathway (b) in blue? This should look familiar - it is simply an SN1 reaction (section 8.1B). In fact, the reaction would result in a mixture of elimination (E1) and substitution (SN1) products. This is a common theme: elimination and substitution often compete with each other.
When both elimination and substitution products are possible, however, we can often predict which reaction will predominate. In general, strong bases and hindered carbons favor elimination, while powerful nucleophiles and unhindered carbons favor substitution.
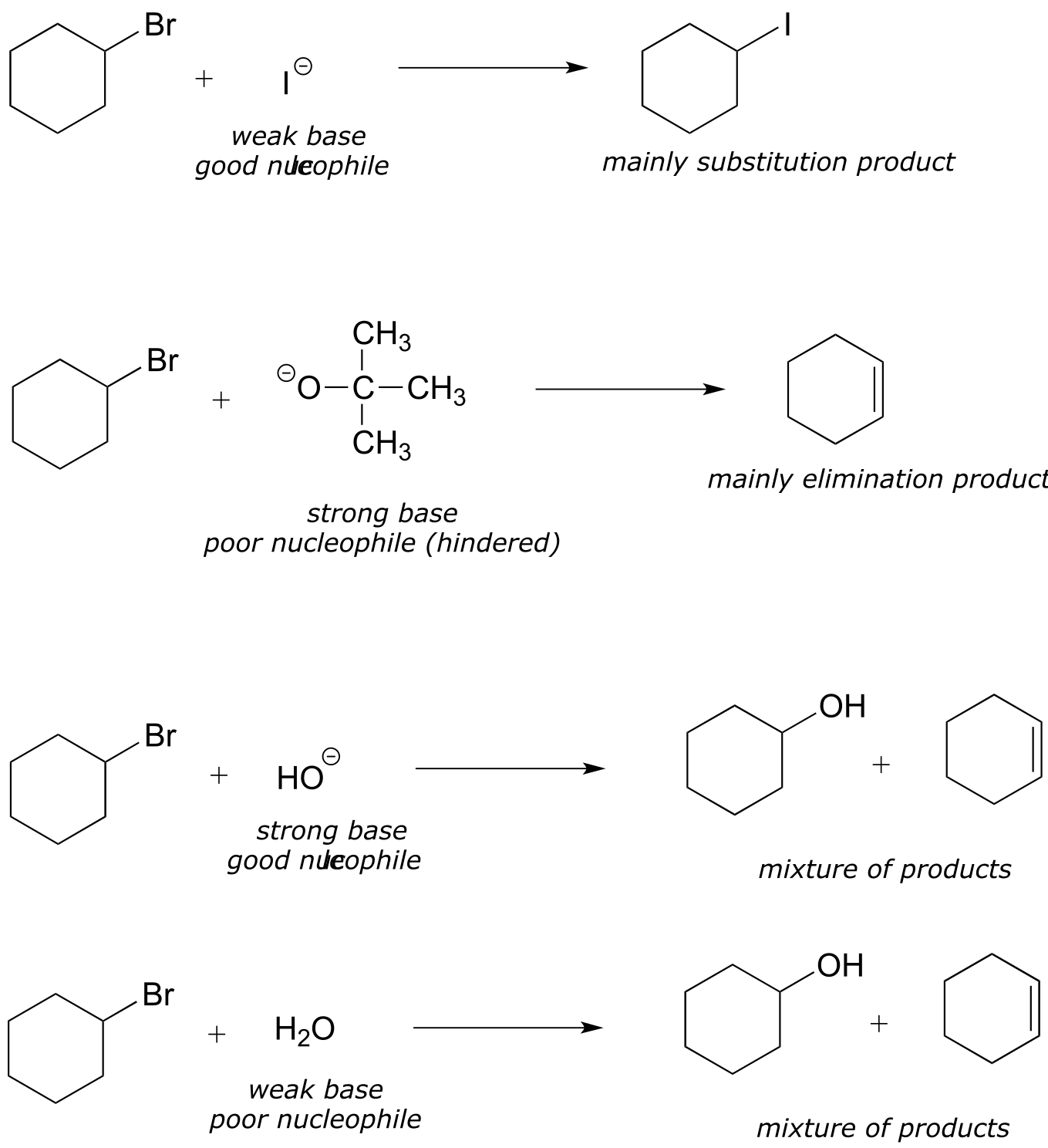
fig 20b
In addition, primary alkyl halides are much more likely to undergo substitution than elimination, even when the nucleophile is also a strong base, because the electrophilic carbon is unhindered and accessible to the nucleophile. Recall that the Williamson ether synthesis (section 8.9A) is an efficient laboratory SN2 reaction between a primary (or methyl) alkyl halide and an alkoxide. If a secondary alkyl halide is used, a substantial amount of elimination product will form (the electrophilic carbon is more hindered, and the alkoxide will act as a base as well as a nucleophile).
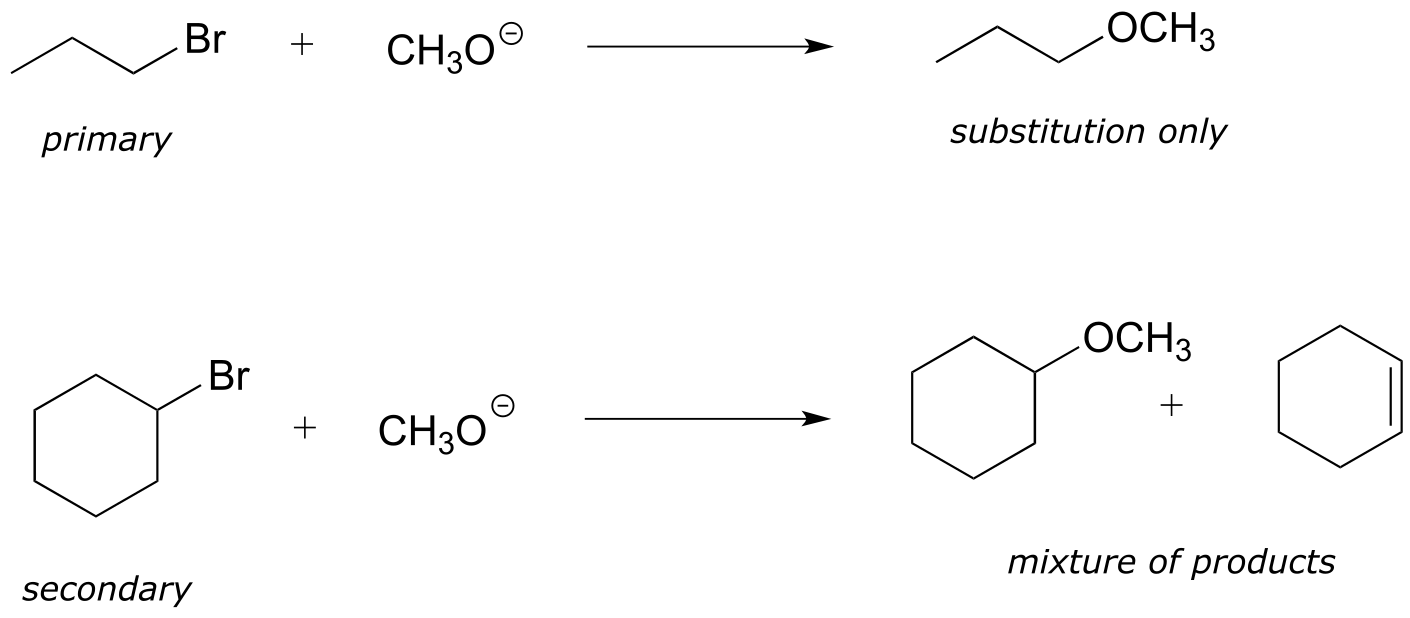
fig 20e
While competition between substitution and elimination pathways is an issue for chemists running reactions in the lab, the same cannot be said of biochemical reactions, as the architecture active site of enzymes have evolved to ensure the formation of only one product.
Exercise 14.9: Predict the major organic product(s) of the following reactions. If the reaction is expected to result in a mixture of elimination and substitution product, show both.
a) bromocyclopentane plus ethoxide
b) 1-chlorohexane plus CH3S-
c) 2-iodo-2-methylpentane plus hydroxide
14.2F: Biochemical E1 elimination reactions#
Looking through metabolic pathways in a biochemistry textbook, you’ll see that almost all elimination reactions appear to be of the E1cb type, occurring on carbons in the β position relative to a carbonyl or imine. A relatively small number of elimination steps, however, take place away from the electron-withdrawing influence of a carbonyl or imine, and these are of the carbocation-intermediate, E1 type. The E2 mechanism is very rare in biochemical pathways.
A reaction in the histidine biosynthetic pathway (EC 4.2.1.19) provides an example of a biological E1 dehydration step:
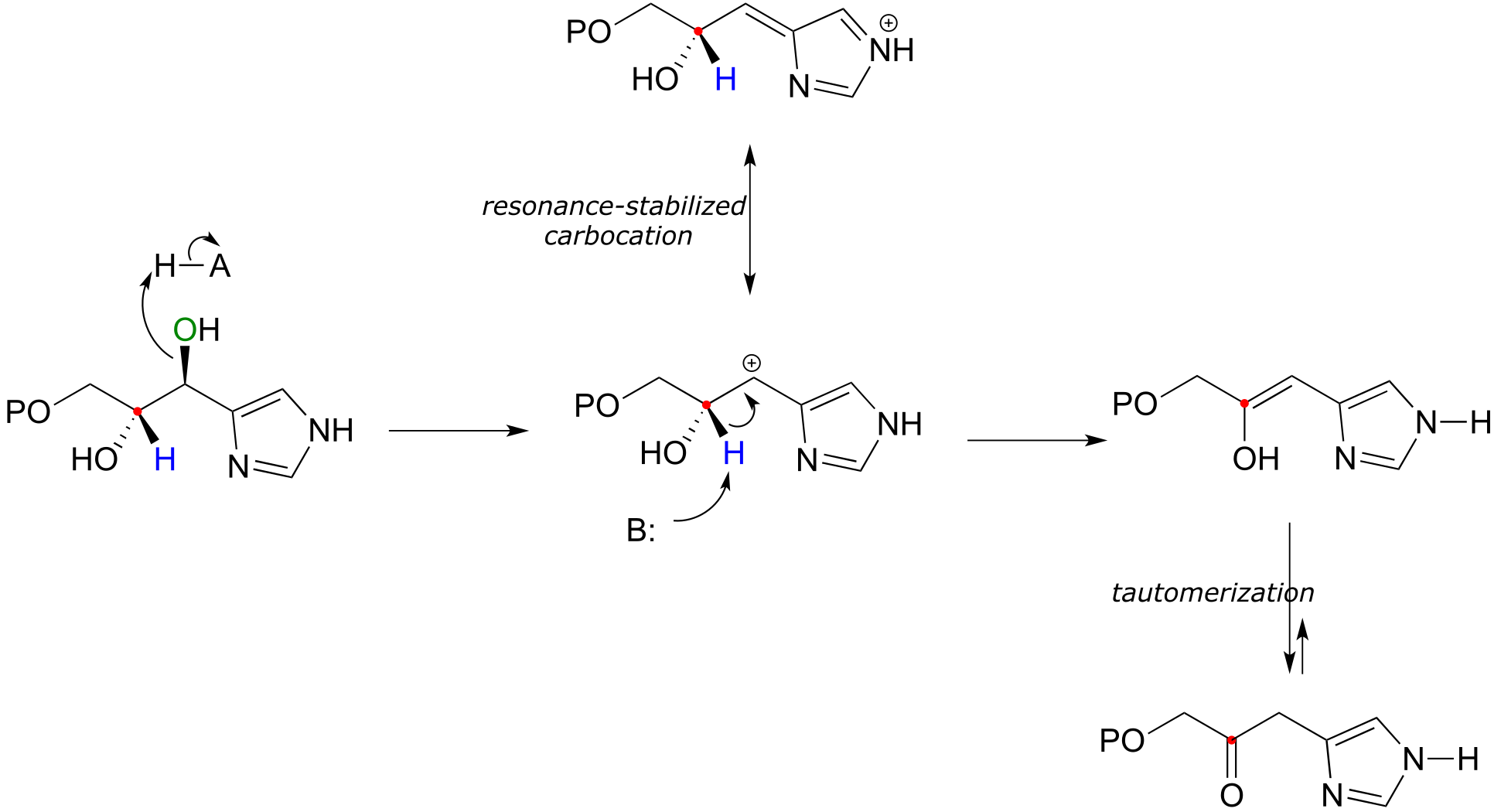
fig 21a
Notice that an E1cb mechanism is not possible here - there is no adjacent carbonyl or imine and thus no possibility for a stabilized anionic intermediate. Instead, the first step is loss of water to form a resonance-stabilized carbocation intermediate. Deprotonation completes the E1 phase of the reaction to form an enol, which rapidly tautomerizes to a ketone.
Another example of a biological E1 reaction is found in the biosynthesis pathway for aromatic amino acids (EC 2.5.1.19):
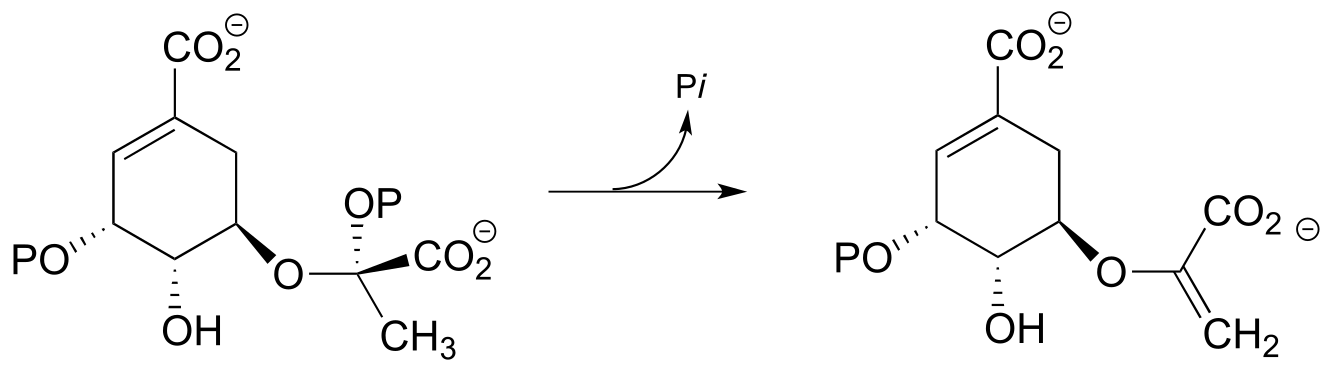
fig 22
Exercise 14.10: Draw a complete mechanism for the reaction above. Show how the carbocation intermediate is stabilized by resonance.
Exercise 14.11 : Another step (EC 4.2.3.5) in the aromatic acid biosynthesis pathway could be referred to as a conjugated E1 elimination of phosphate, the mechanistic reverse of electrophilic addition to a conjugated diene (section 14.1E). Draw a complete mechanism for this reaction, showing two resonance contributors of the carbocation intermediate.
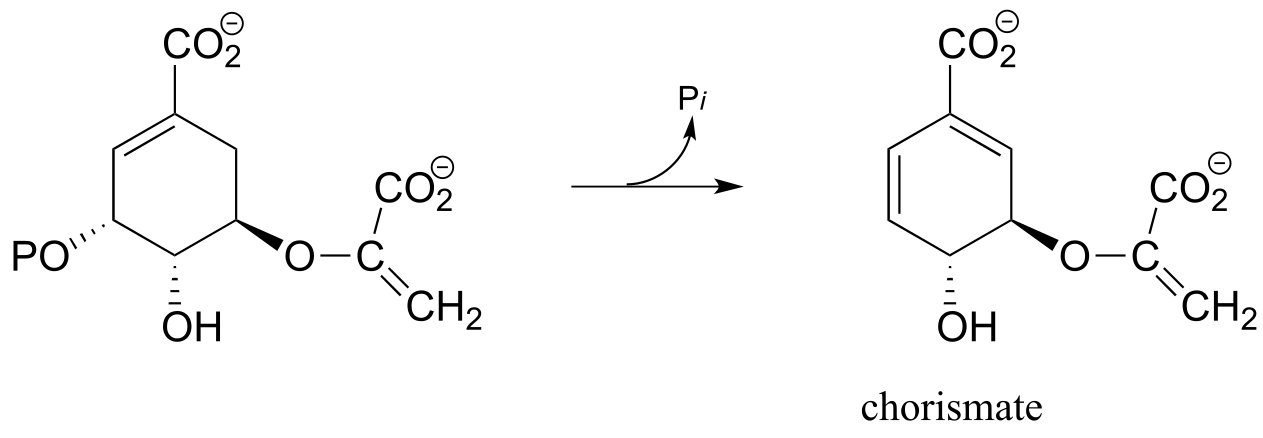
fig 2
In section 13.3, we saw some Claisen condensation reactions in which the usual proton-abstracton step was replaced by decarboxylation. A similar thing can happen with E1 eliminations:
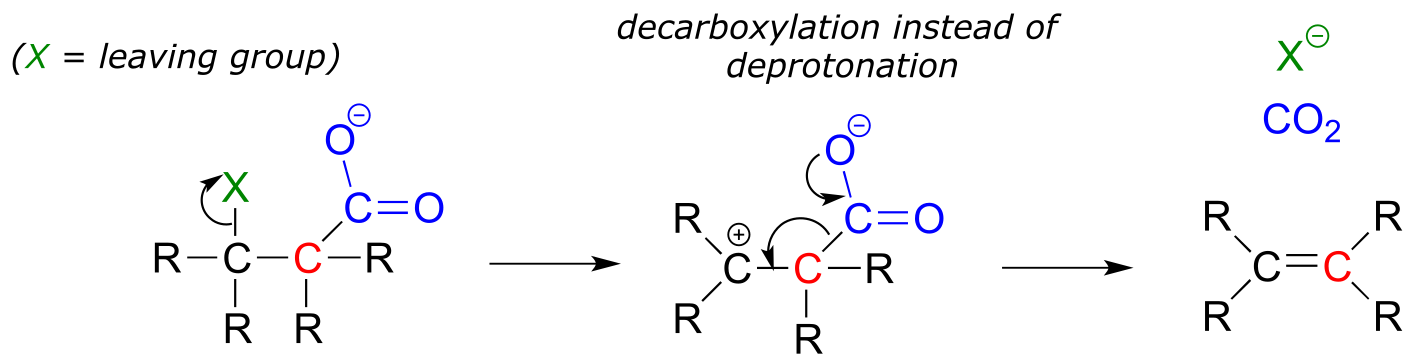
fig 24
Isopentenyl diphosphate, the ‘building block’ for all isoprenoid compounds, is a product of this type of hybrid decarboxylation / elimination reaction (EC 4.1.1.33).
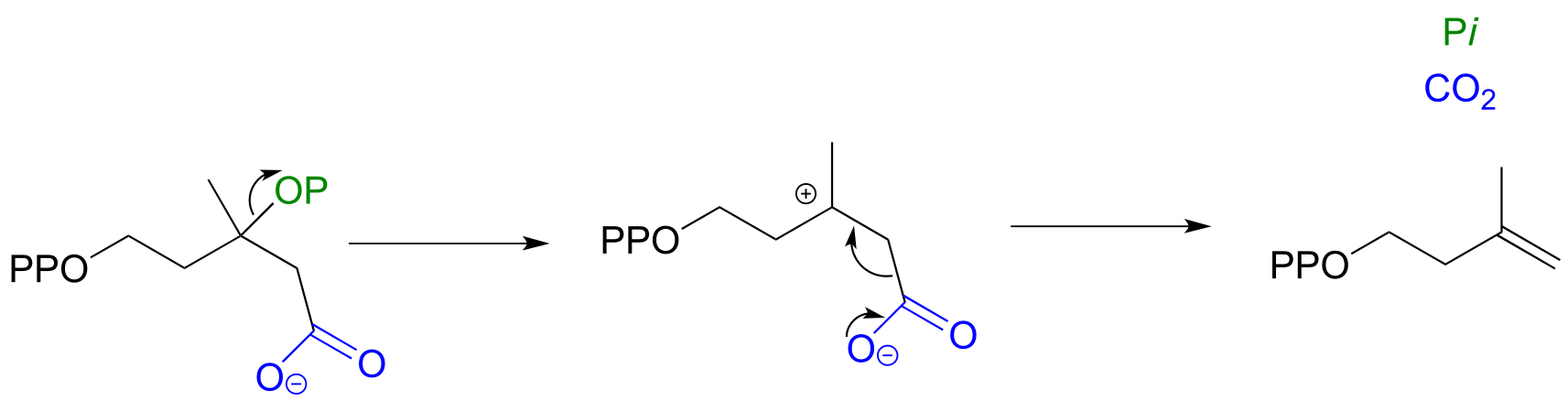
fig 25
Exercise 14.12: A conjugated decarboxylation/E1 elimination reaction (EC 4.2.1.51) occurs in the phenylalanine biosynthesis pathway.
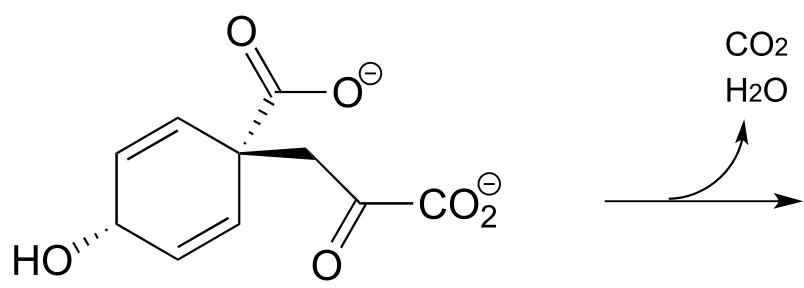
fig 26
a) Predict the product, and draw a mechanism.
b) What two main factors contribute to the ‘driving force’ for this reaction?
Section 14.3: Electrophilic alkene isomeration#
Electrophilic reactions in biochemistry are not limited to addition to alkene double bonds. The position of a double bond in an alkene can also be shifted through an electrophilic, carbocation-intermediate reaction. An electrophilic alkene isomerization occurs when an initial π bond protonation event (step 1 below) is followed by deprotonation of an adjacent carbon to re-form the π bond in a different location.
Electrophilic isomerization mechanism:
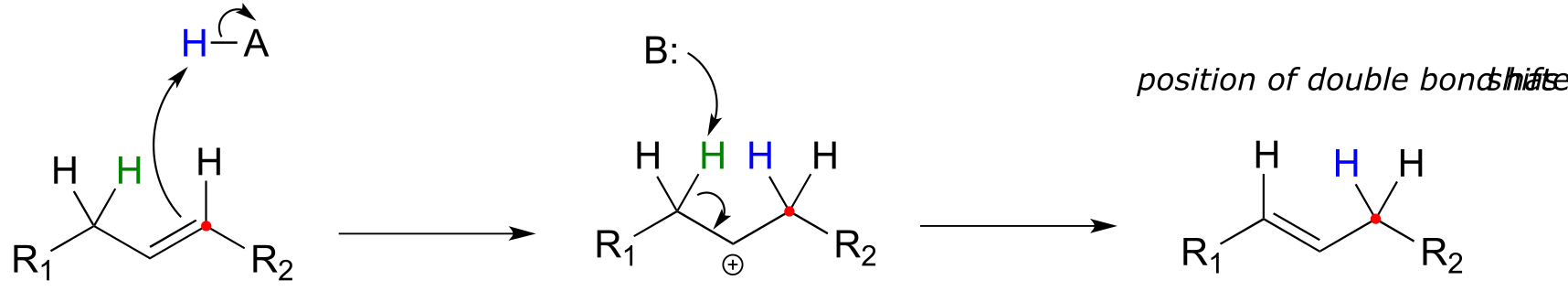
fig 27
In a key early step in the biosynthesis of isoprenoid compounds, isopentenyl diphosphate (IPP), the isoprenoid ‘building block’ molecule, is isomerized to dimethylallyl diphosphate (DMAPP) (EC 5.3.3.2).
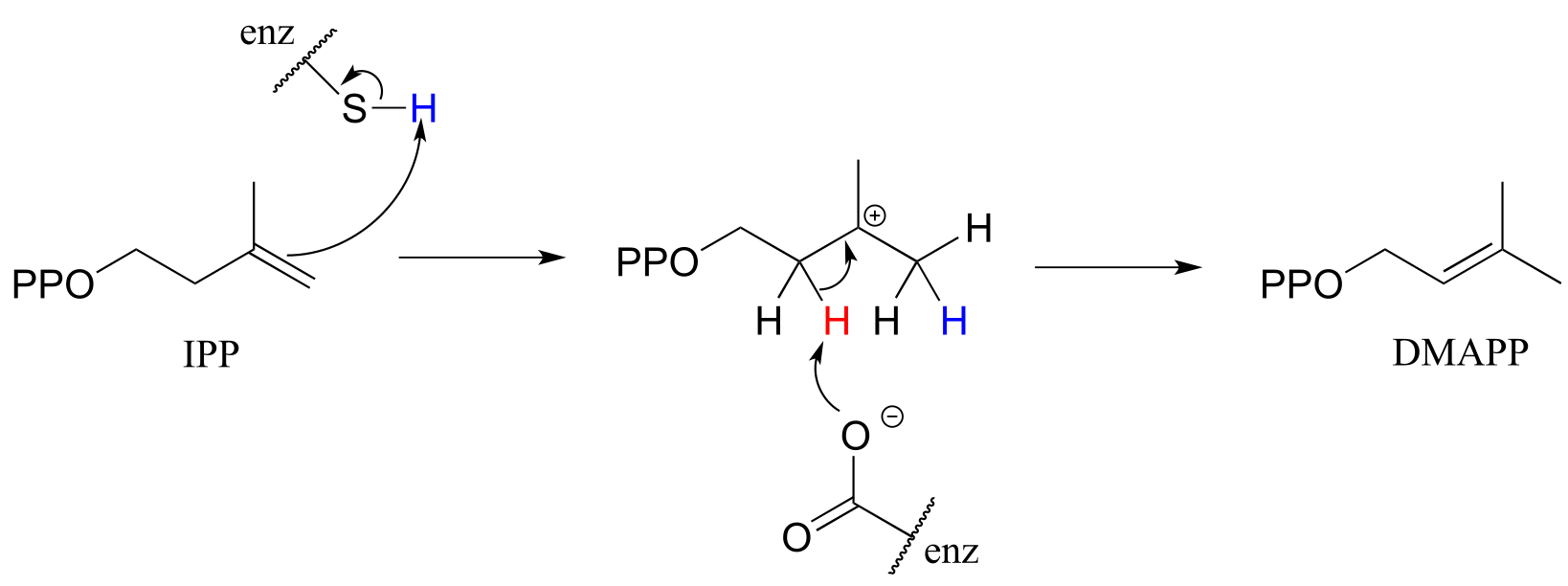
fig 28
In the first step, the π bond between carbon #3 and carbon #4 is protonated by a cysteine residue in the active site. X-ray crystallography studies on the isomerase enzyme (EMBO J. 2001, 20, 1530) show that the carbocation intermediate is bound in a very deep, hydrophobic active site cavity that seals out any water molecules that could potentially attack the carbocation to form an undesired alcohol product. Instead, a basic glutamate residue is positioned in the active site to abstract a proton from carbon #2 (step 2), serving to reestablish the double bond in a new position between carbons #2 and #3.
Section 14.4: Electrophilic substitution#
We have already been introduced to electrophilic addition and electrophilic isomerization - now, let’s move to the third variation on the electrophilic theme, that of electrophilic substitution. In an electrophilic substitution reaction, a pair of π-bonded electrons first attacks an electrophile - usually a carbocation species - and a proton is then abstracted from an adjacent carbon to reestablish the double bond, either in the original position or with isomerization.
Electrophilic substitution mechanism:
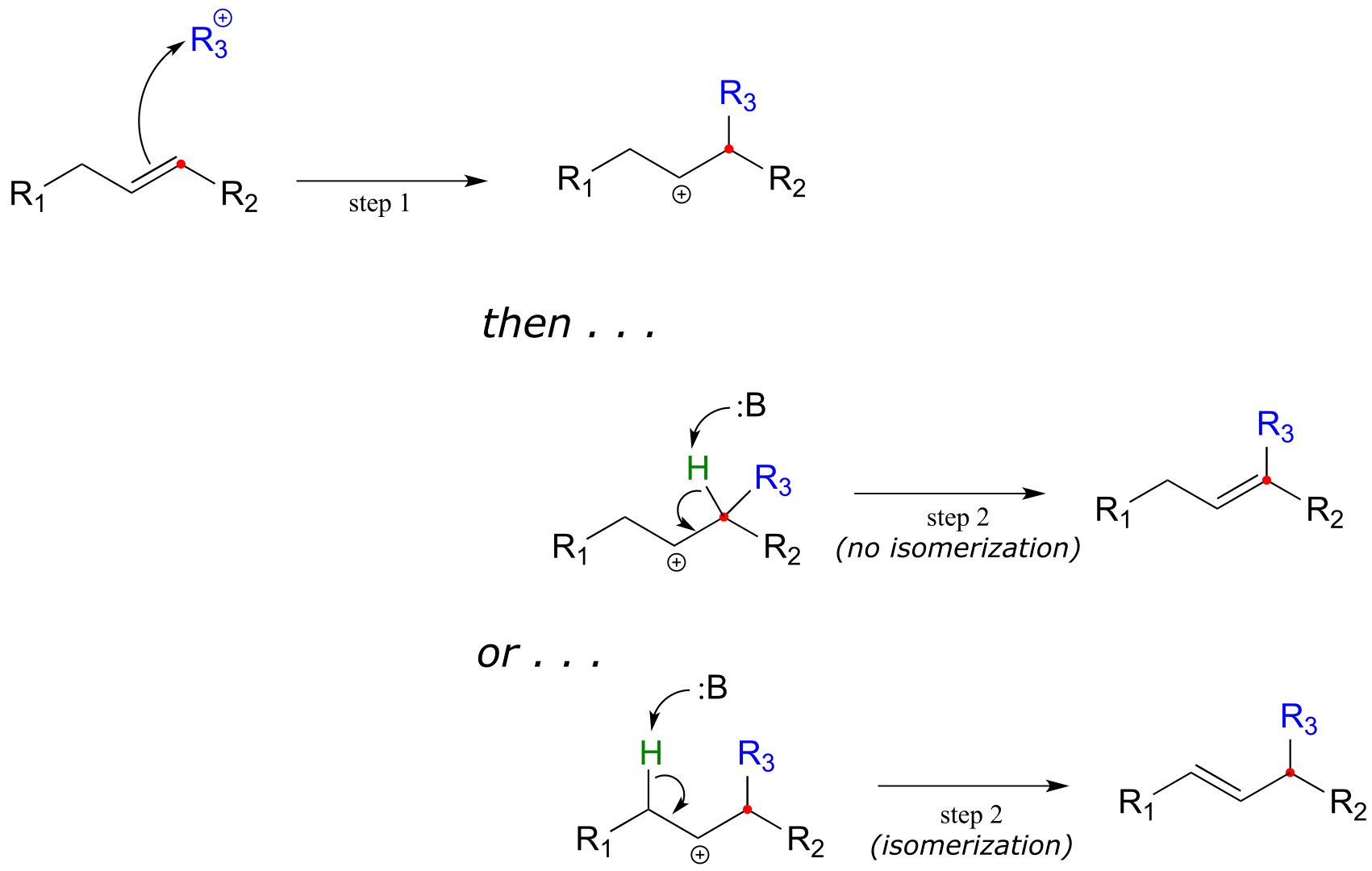
fig 29
14.4A: Electrophilic substitution reactions in isoprenoid biosynthesis#
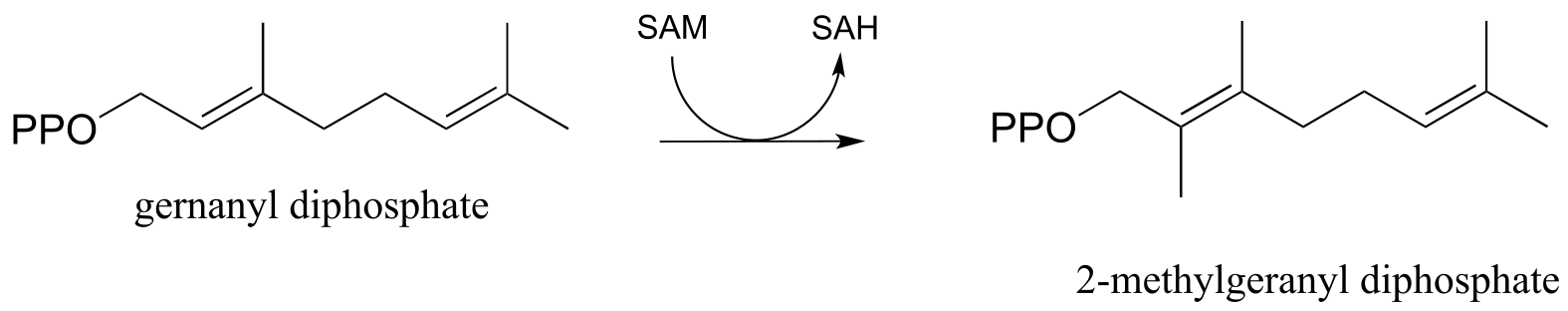
Electrophilic substitution steps are very important in the biosynthetic pathways if isoprenoid compounds. In an early, chain-elongating reaction (EC 2.5.1.1) of the pathways of many isoprenoids, building blocks IPP and DMAPP combine to form a 10-carbon isoprenoid product called geranyl diphosphate (GPP):
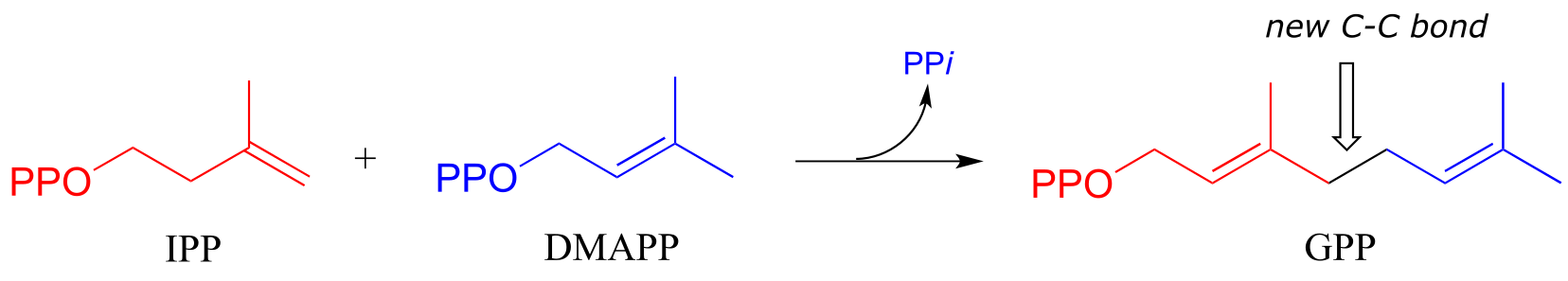
fig 30
In a preliminary step (step a below), the diphosphate group on DMAPP departs to form an allylic carbocation.
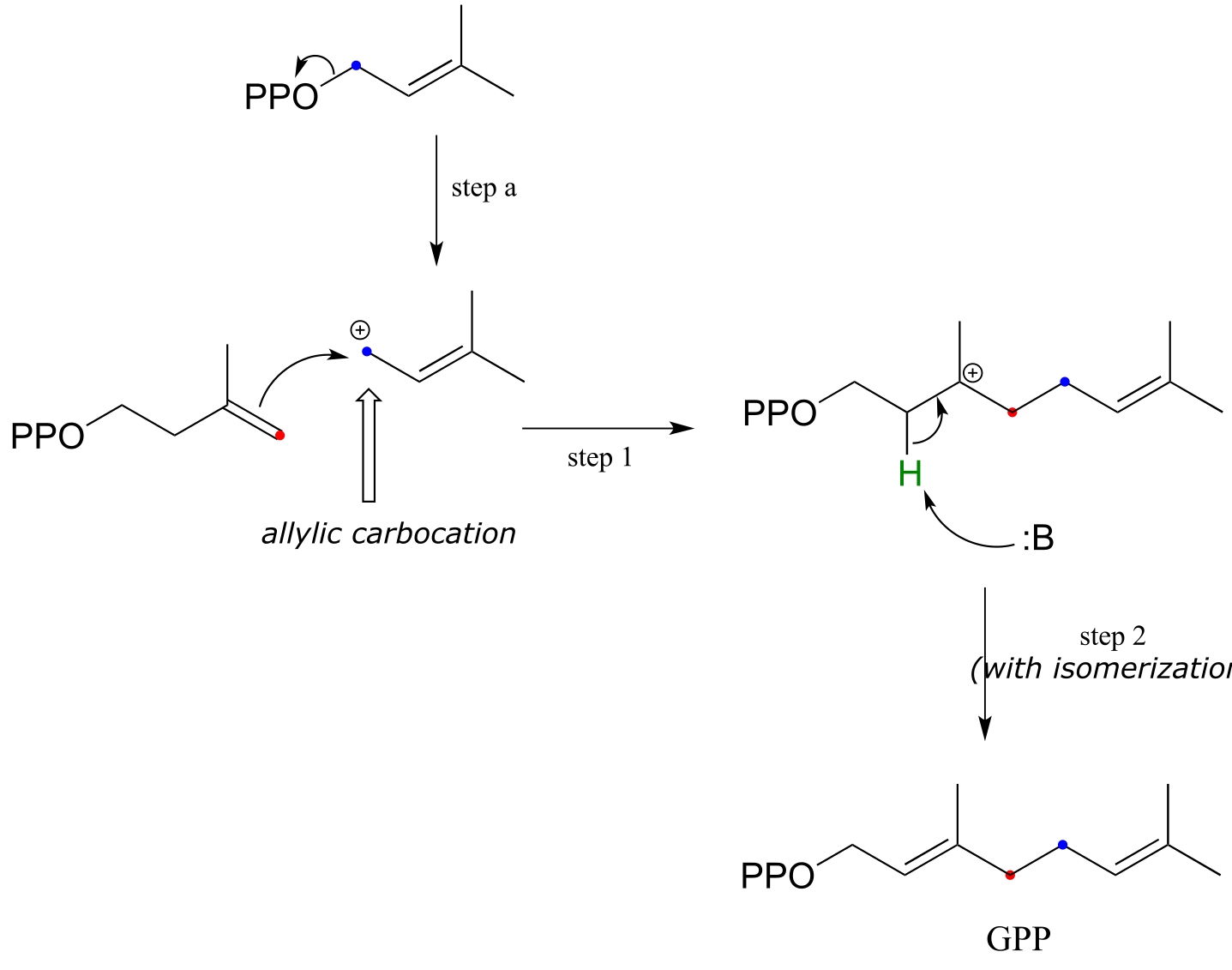
fig 32
In step 1, the π electrons in IPP then attack the electrophilic carbocation from step a, resulting in a new carbon-carbon bond and a tertiary carbocation intermediate. Proton abstraction (step 2) leads to re-establishment of a double bond one carbon over from where it started out in IPP.
Exercise 14.13: DMAPP is much more prone to spontaneous hydrolysis than IPP when they are dissolved in water. Explain why.
Exercise 14.14: Farnesyl diphosphate (FPP) is synthesized by adding another five-carbon building block to geranyl diphosphate. What is this building block - IPP or DMAPP? Draw a mechanism for the formation of FPP.
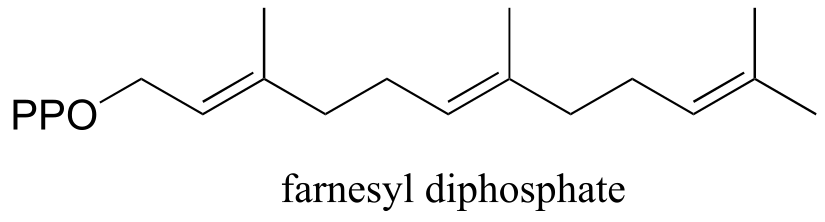
fig 33a
Exercise 14.15: Propose a likely mechanism for the following transformation, which is the first stage in a somewhat complex reaction in the synthesis of an isoprenoid compound in plants. (Science 1997, 277, 1815)
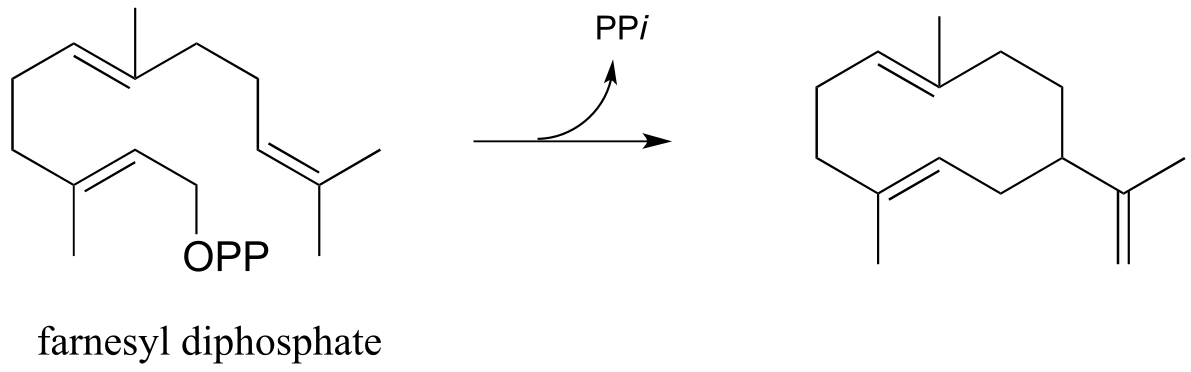
fig 33
Exercise 14.16: The electrophilic carbon in an electrophilic substitution reaction is often a carbocation, but it can also be the methyl group on S-adenosylmethionine (SAM - see section 8.8A). Propose a likely mechanism for this methylation reaction. (Biochemistry 2012, 51, 3003)
14.4B: Electrophilic aromatic substitution#
Until now, we have been focusing mostly on electrophilic reactions of alkenes. Recall from section 2.2C that π bonds in aromatic rings are substantially less reactive than those in alkenes. Aromatic systems, however, do in fact undergo electrophilic substitution reactions given a powerful electrophile such as a carbocation, and if the carbocation intermediate that forms can be sufficiently stabilized.
Electrophilic aromatic substitution (Friedel-Crafts alkylation) mechanism
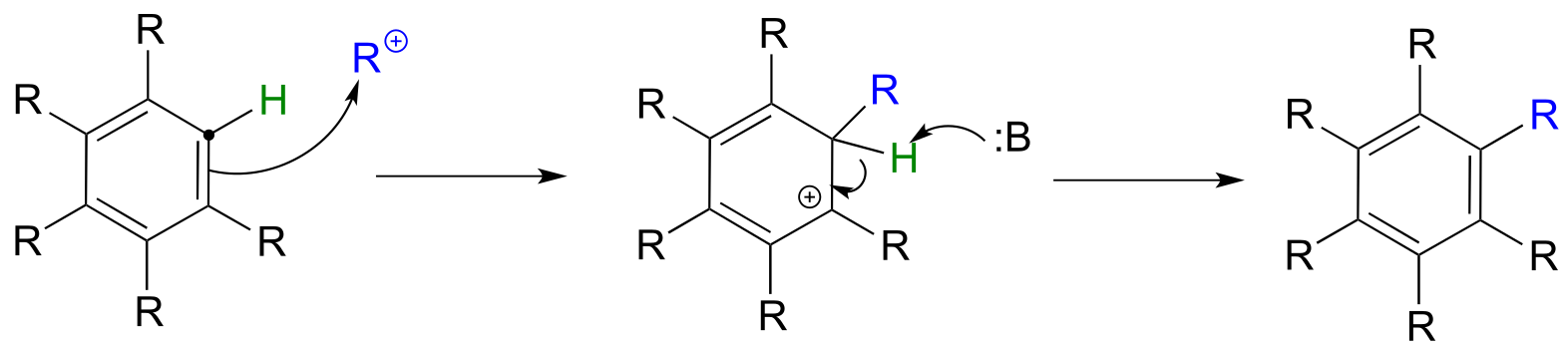
fig 34
Organic chemists often refer to electrophilic aromatic substitution reactions with carbocation electrophiles as Friedel-Crafts alkylation reactions.
Exercise 14.17: Aromatic rings generally do not undergo electrophilic addition reactions. Why not?
The Friedel-Crafts reaction below is part of the biosynthesis of vitamin K and related biomolecules.
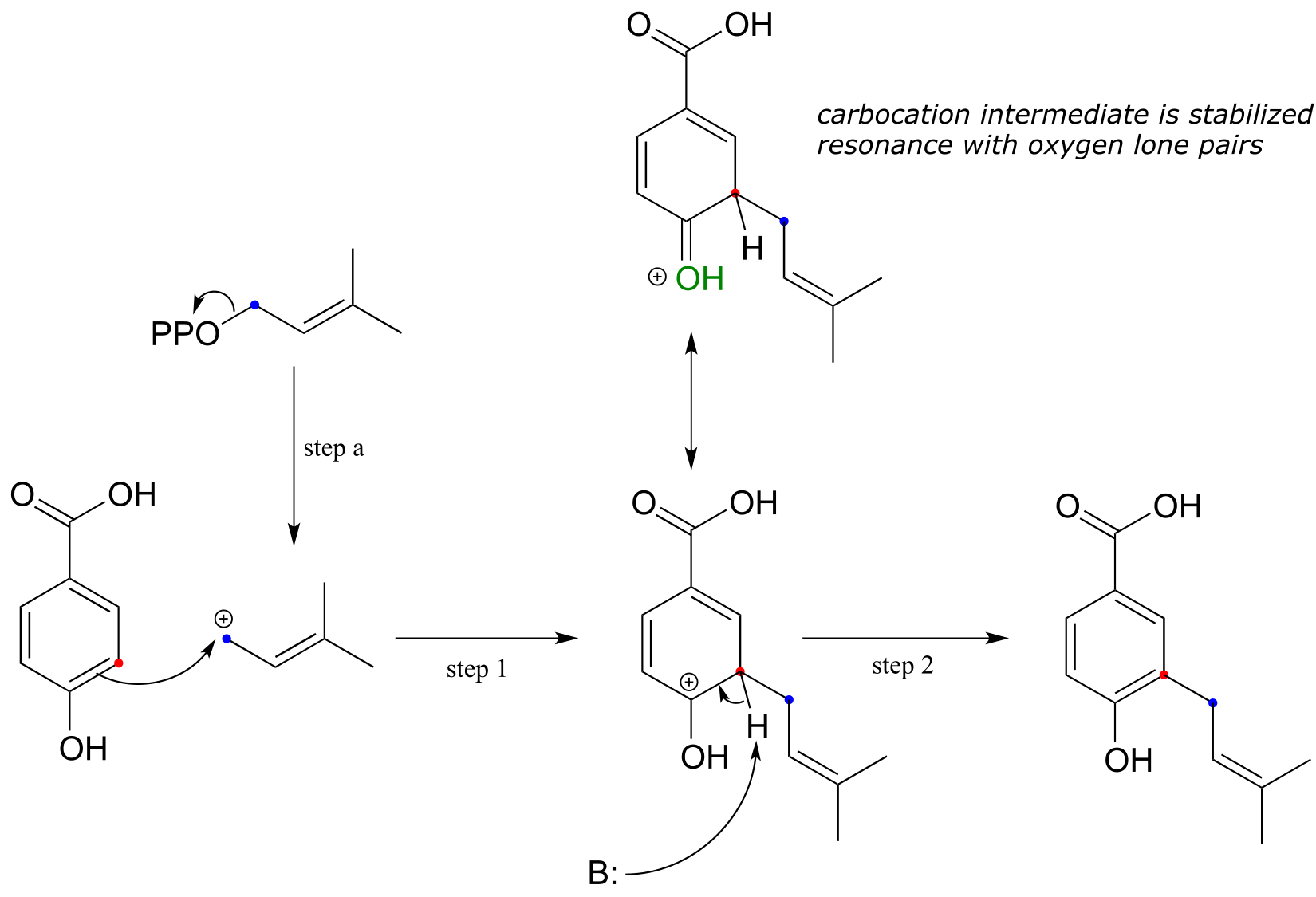
fig 36
Loss of diphosphate creates a powerful carbocation electrophile (step a) which attracts the π electrons of the aromatic ring to form a carbocation intermediate with a new carbon-carbon bond (step 1). Substitution is completed by proton abstraction (step 2) which re-establishes the aromatic sextet.
An important point must be made here: because aromatic π bonds are substantially less reactive than alkene π bonds, the electrophilic must be VERY electrophilic - usually a carbocation. In addition, the carbocation intermediate that results from attack by aromatic π electrons is generally stabilized by resonance with lone pair electrons on a nearby oxygen or nitrogen (look at the resonance forms of the positively-charged intermediate that forms as the result of step 1 in the above figure).
Remember that stabilizing the intermediate formed in a rate-limiting step has the effect of lowering the activation energy for the step, and thus accelerating the reaction.
Organic chemists use the term ring activation to refer to the rate-accelerating effect of electron-donating heteroatoms in electrophilic aromatic substitution reactions. Aromatic rings lacking any activating oxygen or nitrogen atoms are less reactive towards electrophilic substitution.
An example of the ring-activating effect of the nitrogen atom on an aromatic ring can be found in the following Friedel-Crafts reaction (EC 2.5.1.34), which should be familiar from the introduction to this chapter:
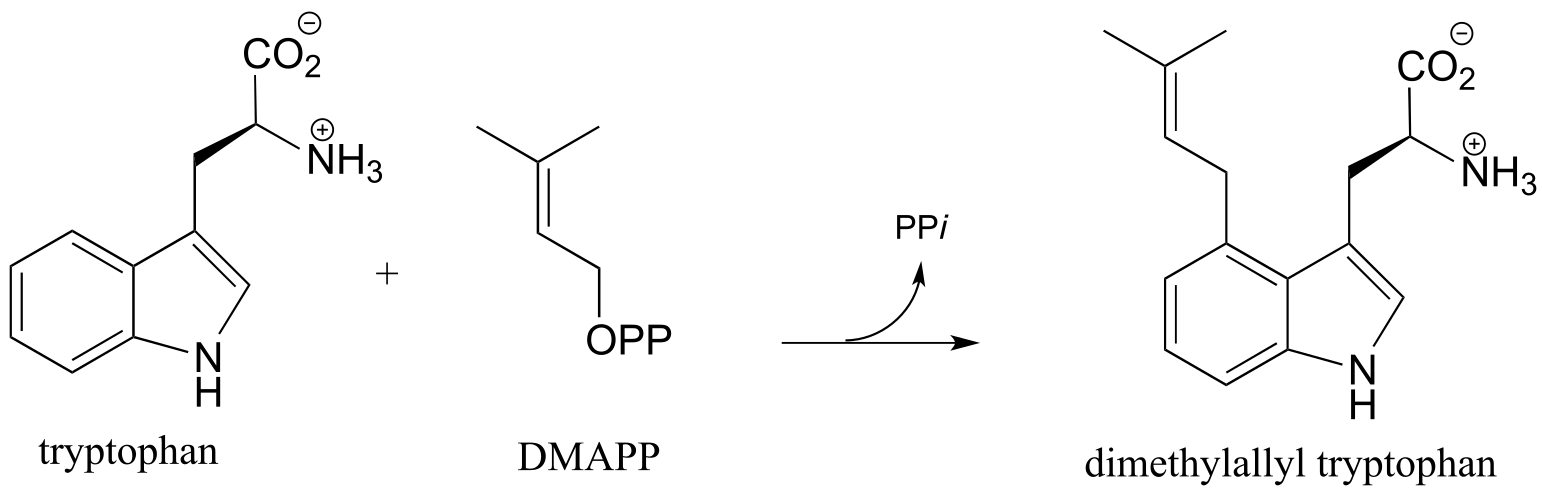
fig 37
Recall that this is a key early step in the biosynthetic pathway for the ergot alkaloids which are hypothesized to have been the root cause of the ‘bewitchment’ of several young girls in 17th century Salem, Massachusetts. (J. Am. Chem. Soc. 1992*,114,* 7354).
Exercise 14.18: Draw a likely mechanism for the biosynthesis of dimethylallyl tryptophan, including a resonance structure showing how the carbocation intermediate in the rate determining step is stabilized by lone pair electrons on the ring nitrogen (in other words, show how the nitrogen serves to activate the ring).
Friedel-Crafts reactions, in addition to being important biochemical transformations, are commonly carried out in the laboratory. It is instructive to consider a few examples to see how the same principles of structure and reactivity apply to both biochemical and laboratory reactions.
Below is an example of a laboratory Friedel-Crafts alkylation reaction:
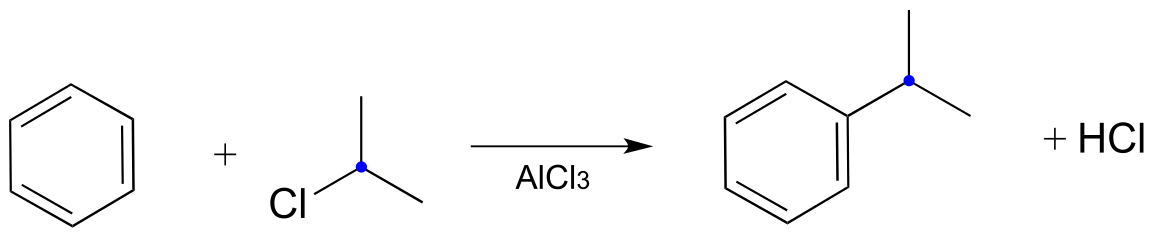
fig 37a
Recall that a powerful electrophile - such as a carbocation - is required for an electrophilic aromatic substitution to occur. The 2-chloropropane reactant is electrophilic, but not electrophilic enough to react with benzene. Here’s where the aluminum trichloride catalyst comes in: it reacts as a Lewis acid with the alkyl chloride to generate a secondary carbocation:
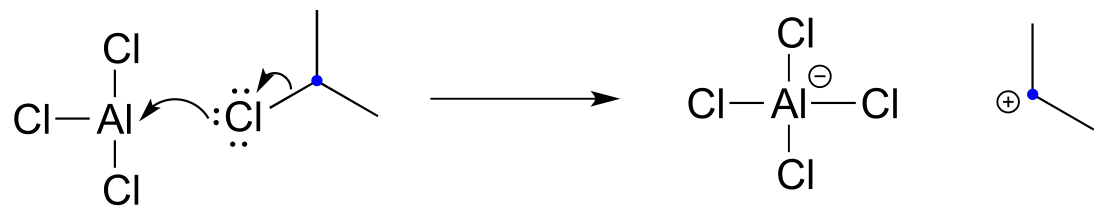
fig 37b
The carbocation thus generated is sufficiently electrophilic to react with the aromatic π electrons, in a manner that should be familiar from the biochemical examples discussed above:

fig 37c
You may have noticed, however, that one element from the biochemical Friedel-Crafts reactions is missing here: there is no activating group to stabilize the ring carbocation intermediate. Indeed, the presence of an activating group - for example, the oxygen atom of a methoxy substituent - greatly increases the rate of a Friedel-Crafts alkylation.
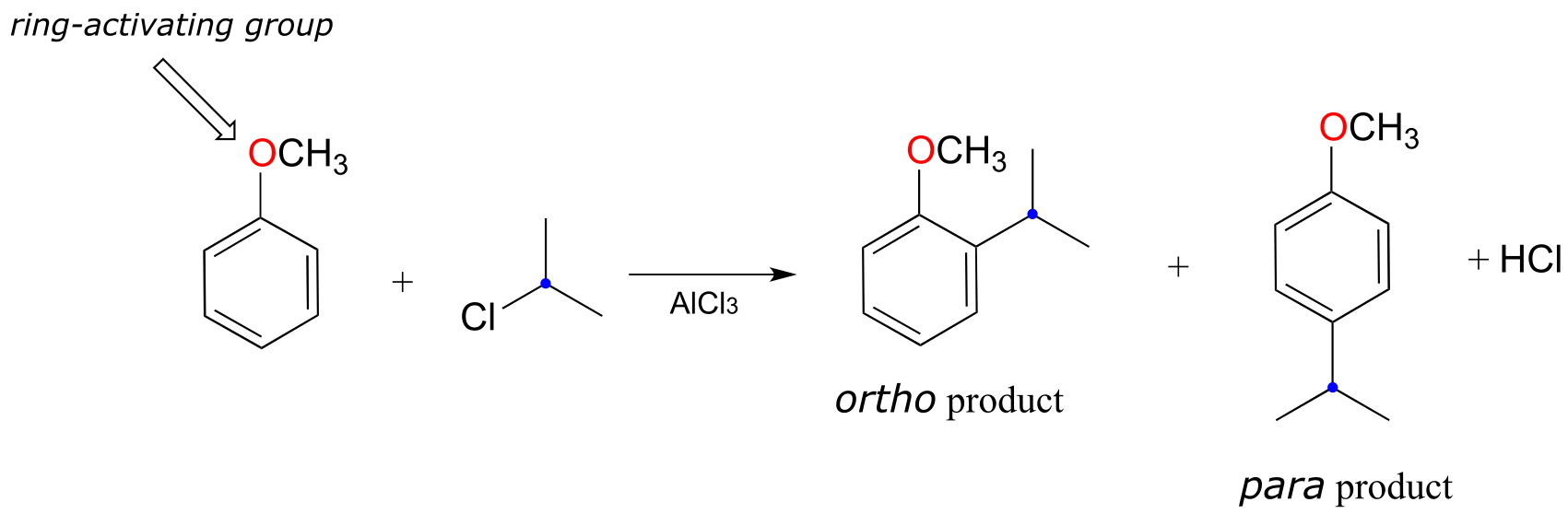
fig 37d
Note in the example shown above that two products are formed: one is an ortho-disubstituted benzene and one is para-disubstituted. Note also that no meta-disubstituted product is formed. This phenomenon is referred to as the ortho-para directing effect, and you are led towards an explanation in the exercise below.
Exercise 14.19:
a) Draw the lowest-energy resonance contributors of the carbocation intermediates leading to formation of the ortho and para products in the reaction above. Use resonance structures to illustrate how the methoxy substituent is a ring-activating group.
b) Draw the hypothetical carbocation intermediate in a reaction leading to formation of a meta-disubstituted product. Is this carbocation stabilized by the methoxy oxygen? Can you see why no meta product forms?
Exercise 14.20:
a) Just as there are ring-activating groups in electrophilic aromatic substitutions, there are also ring-deactivating groups. For each of the substituted benzene reactants below, draw the carbocation intermediate leading to the ortho substitution product and decide whether the substituent is ring-activating or ring-deactivating in a Friedel-Crafts reaction with 2-chloropropane and AlCl3 (in other words, which compounds would react faster than benzene, and which would react slower?) Explain how the ring-deactivating effect works.
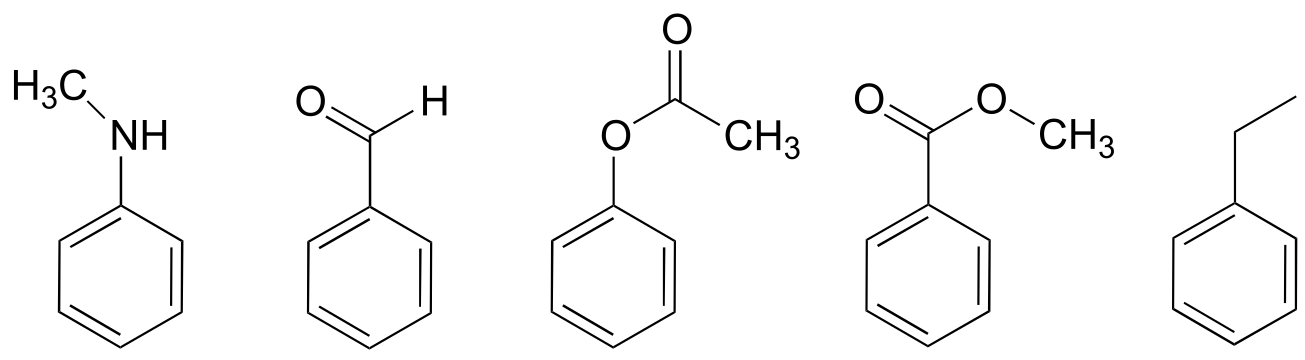
b) (challenging!) Ring-deactivating substituents are usually also meta-directing. Use one of your carbocation intermediate drawings from part (a) of this exercise, and the concept od resonance, to explain this observation.
c) (answer part (b) first) Look again at the vitamin K biosynthesis reaction, and discuss the ring activating/directing effects of the two substituents on the substrate.
Section 14.5: Carbocation rearrangements#
Earlier in this chapter we introduced the so-called ‘Markovnikov rule’, which can be used to predict the favored regiochemical outcome of electrophilic additions to asymmetric alkenes. According to what we have learned, addition of HBr to 3-methyl-1-butene should result in a secondary bromoalkane. However, the predominant product that is actually be observed in this reaction is a tertiary alkyl bromide! Little or no secondary alkyl bromide forms.
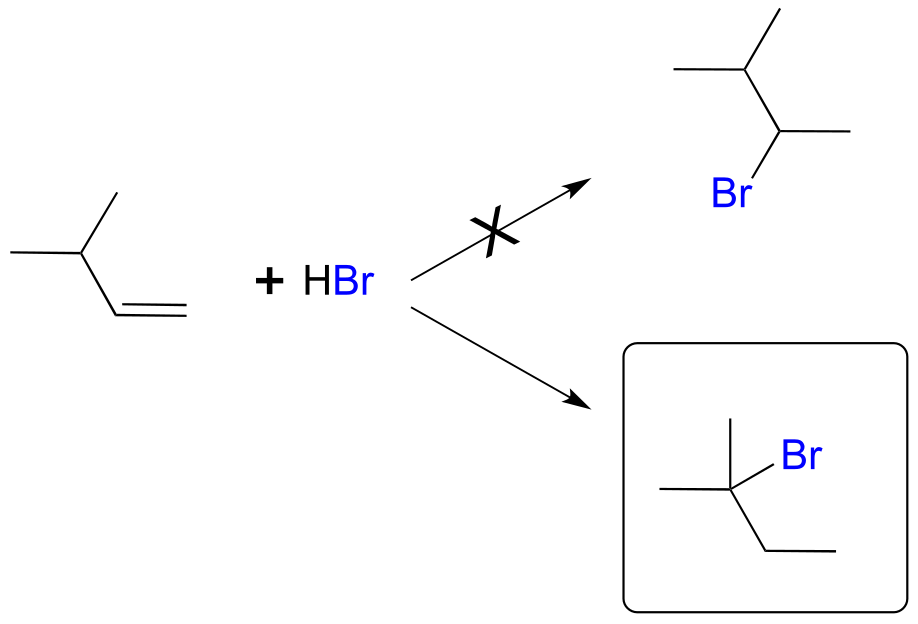
fig 38
To explain this result, let’s take a look at the mechanism for the reaction:
Electrophilic addition with a hydride shift:
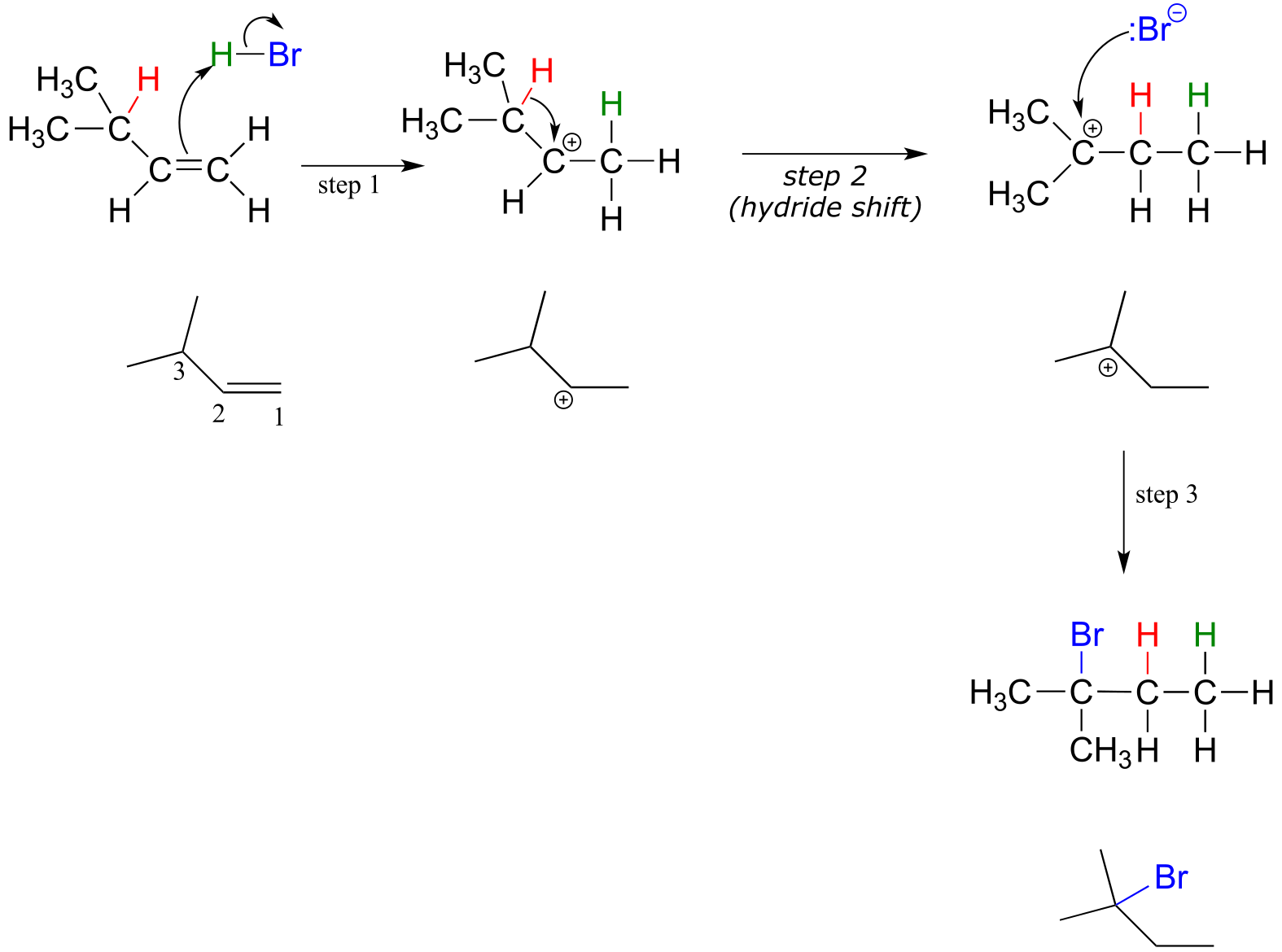
fig 39
Protonation of the double bond results in a secondary carbocation (step 1). What happens next (step 2 above) is a process called a carbocation rearrangement, and more specifically, a hydride shift. The electrons in the bond between carbon #3 and a hydrogen are attracted by the positive charge on carbon #2, and they simply shift over to fill the empty p orbital, pulling the proton over with them. Notice that the hydride, in shifting, is not acting as an actual leaving group - a hydride ion is a very strong base and a very poor leaving group.
An important reminder: a hydride ion (H-) is a proton plus two electrons. Be sure not to confuse a hydride ion with H+, which is just a proton without any electrons.
As the shift proceeds, a new C-H σ bond is formed at carbon #2, and carbon #3 is left with an empty p orbital and a positive charge.
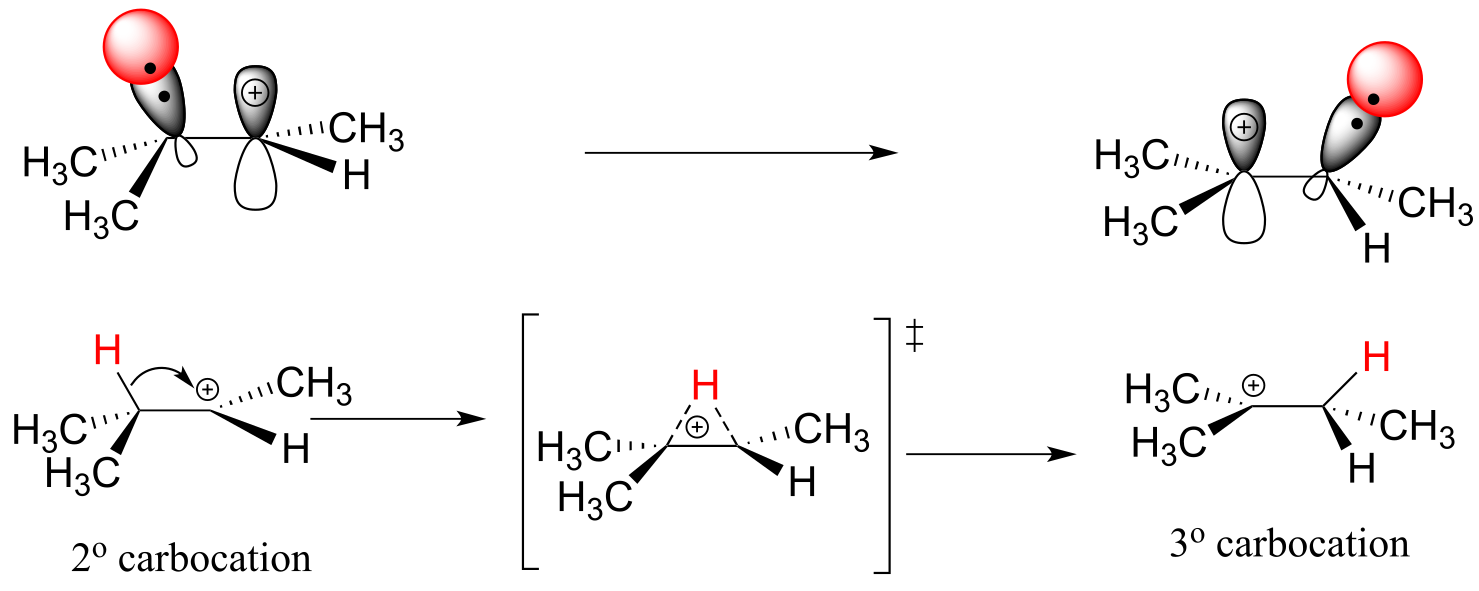
fig 40
What is the thermodynamic driving force for this process? Notice that the hydride shift results in the conversion of a secondary carbocation to a (more stable) tertiary carbocation - a thermodynamically downhill step. As it turns out, the shift occurs so quickly that it is accomplished before the bromide nucleophile has time to attack at carbon #2. Rather, the bromide will attack (step 3) at carbon #3 to complete the addition.
Consider another example. When HBr is added to 3,3-dimethyl-1-butene, the product is a tertiary - rather than a secondary - alkyl bromide.
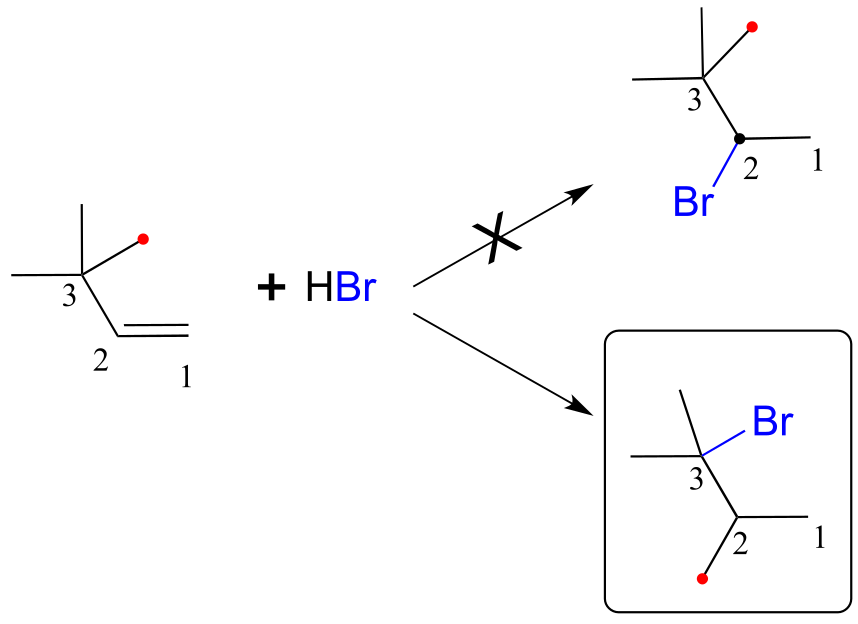
fig 41
Notice that in the observed product, the carbon framework has been rearranged: the methyl carbon indicated by a red dot has shifted from carbon #3 to carbon #2. This is an example of another type of carbocation rearrangement, called a methyl shift.
Below is the mechanism for the reaction. Once again a secondary carbocation intermediate is formed in step 1. In this case, there is no hydrogen on carbon #3 available to shift over create a more stable tertiary carbocation. Instead, it is a methyl group that does the shifting, as the electrons in the carbon-carbon σ bond move over to fill the empty orbital on carbon #2 (step 2 below).
Electrophilic addition with methyl shift:
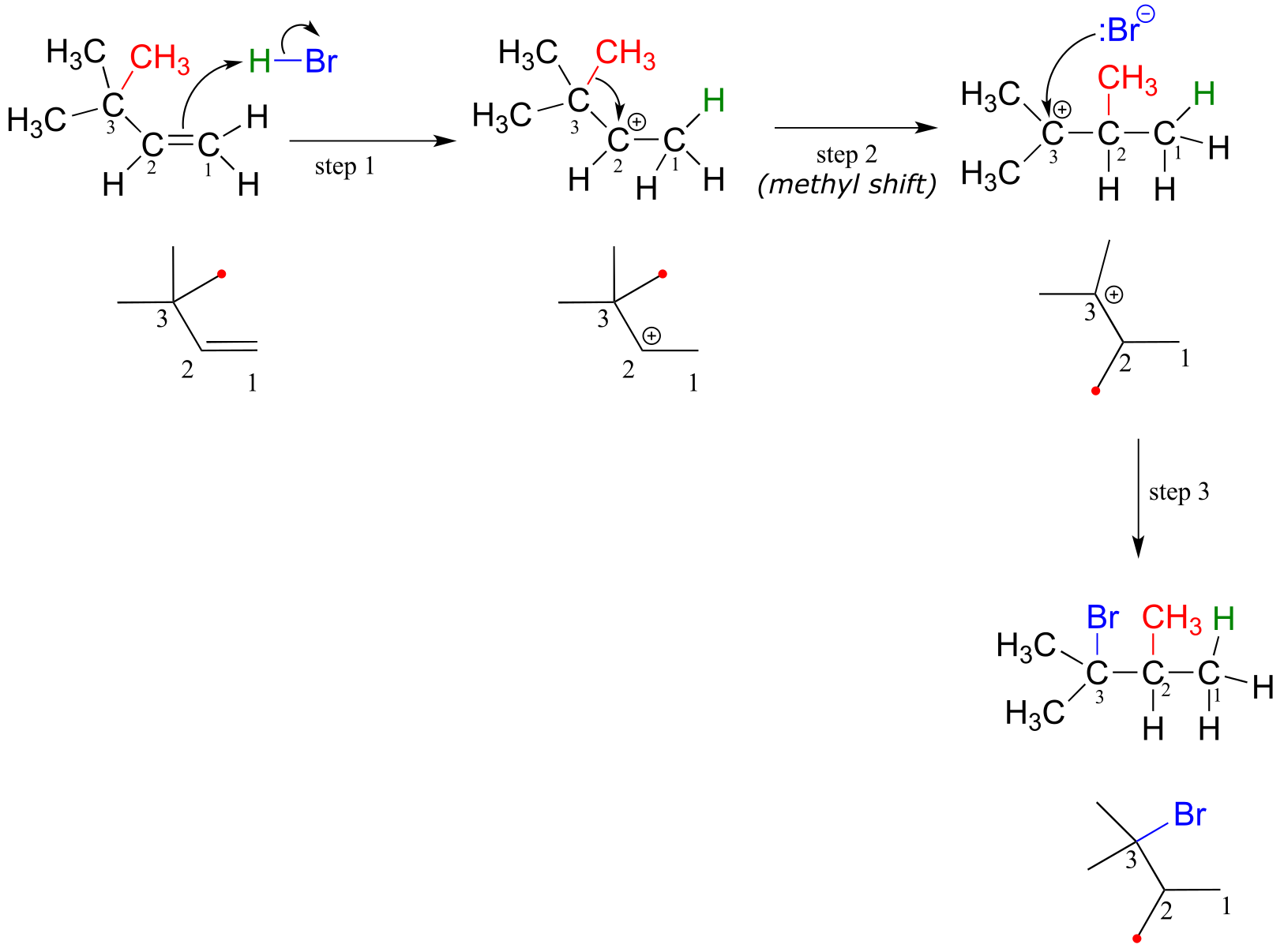
fig 42
The methyl shift results in the conversion of a secondary carbocation to a more stable tertiary carbocation. The end result is a rearrangement of the carbon framework of the molecule.
Exercise 14.21: Which of the following carbocations are likely to undergo a shift? If a shift is likely, draw the new carbocation that would result.
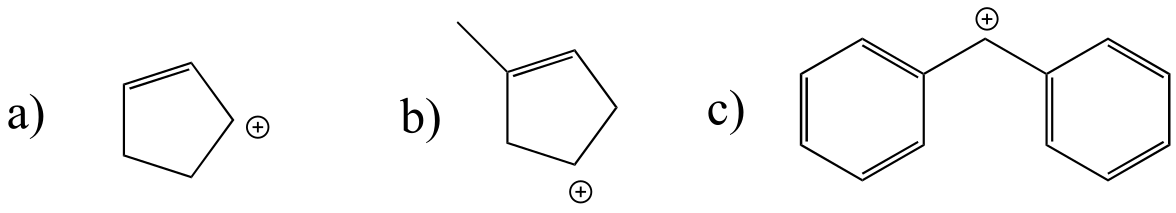
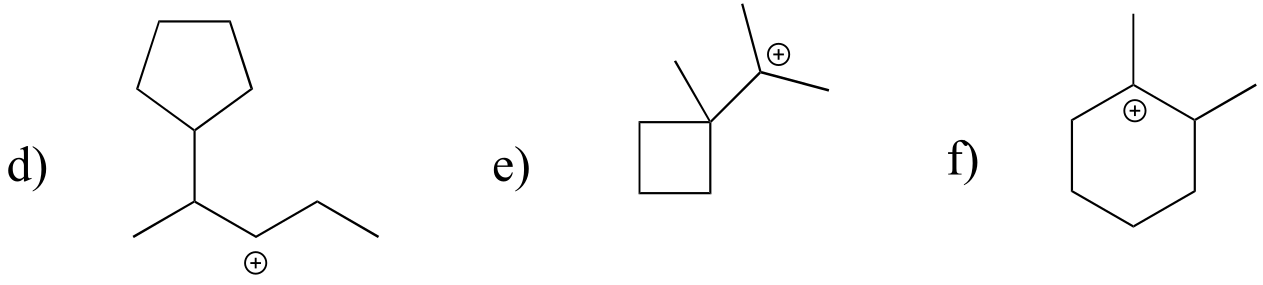
Exercise 14.22: In the (non-biochemcial) reactions below, the major product forms as the result of a hydride or methyl shift from a carbocation intermediate. Predict the structure of the major product for each reaction, disregarding stereochemistry.
a)
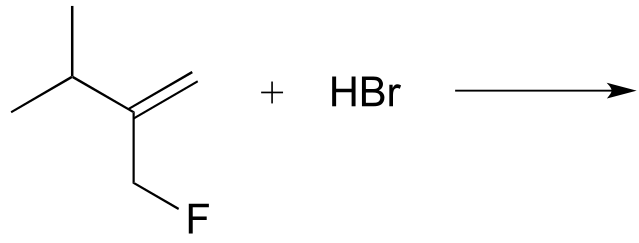
b)
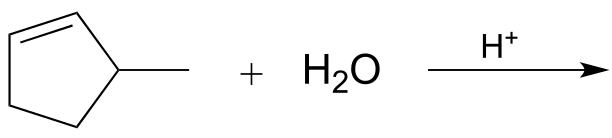
c)
d)
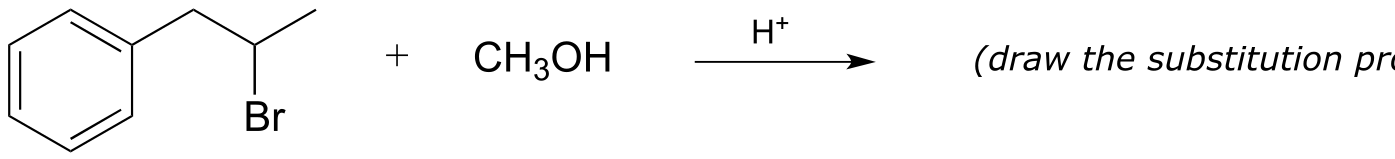
fig 43
Exercise 14.23: Draw the most abundant product of this laboratory Friedel-Crafts reaction:
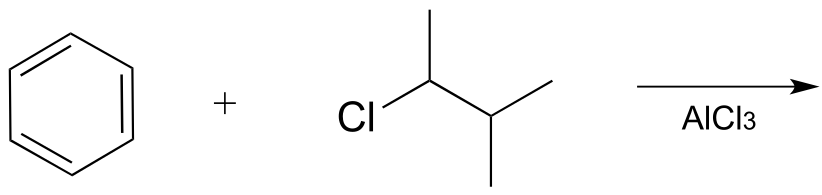
fig 43a
Carbocation rearrangements are involved in many known biochemical reactions. Rearrangements are particularly important in carbocation-intermediate reactions in which isoprenoid molecules cyclize to form complex multi-ring structures. For example, one of the key steps in the biosynthesis of cholesterol is the electrophilic cyclization of oxidosqualene to form a steroid called lanosterol (E.C. 5.4.99.7).
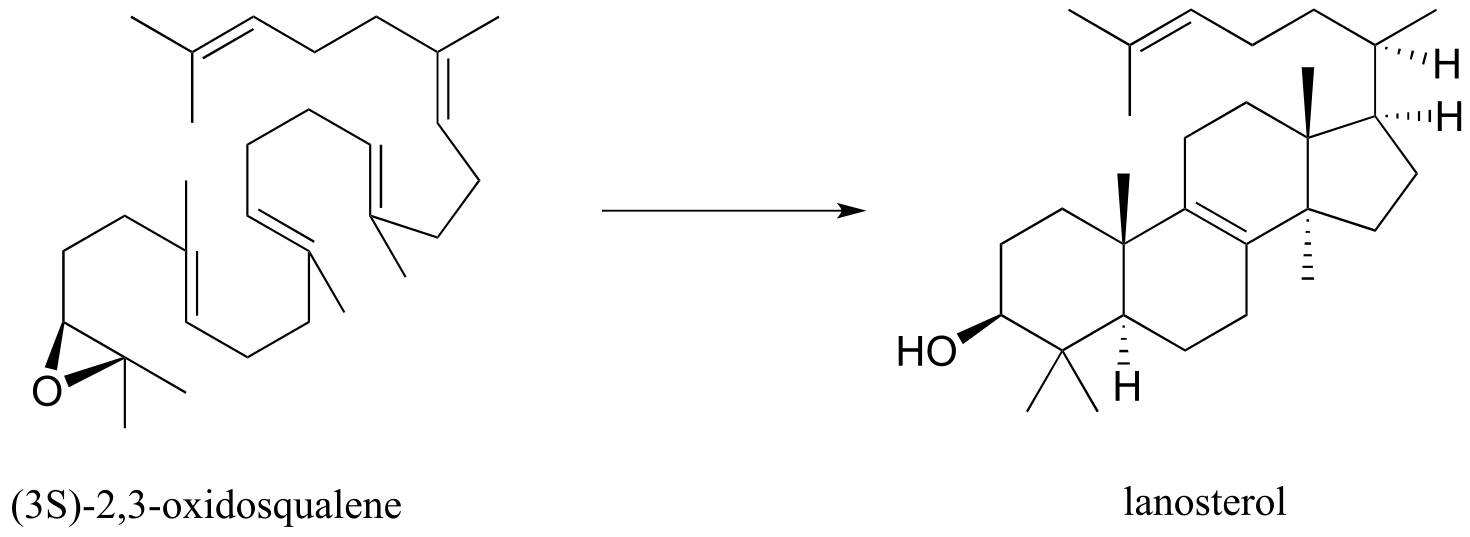
fig 44
This complex but fascinating reaction has two phases. The first phase is where the actual cyclization takes place, with the formation of four new carbon-carbon bonds and a carbocation intermediate. This phase is a ‘cascade’ of electrophilic alkene addition steps, beginning with addition of an electrophilic functional group called an ‘epoxide’.
The epoxide functional group - composed of a three membered ring with two carbons and an oxygen - is relatively rare in biomolecules and biochemical reactions, and for this reason it is not discussed in detail in this book. However, epoxides are an important and versatile intermediate in laboratory organic synthesis, so you will learn much more about how they are made and how they react if you take a course in chemical synthesis. For now, it is sufficient to recognize that the carbon atoms of an epoxide are potent electrophiles, due to both the carbon-oxygen bond dipoles and the inherent strain of the three membered ring.
The second phase involves a series of hydride and methyl shifts culminating in a deprotonation. In the exercise below, you will have the opportunity to work through the entire cyclase reaction mechanism. In section 15.7, we will take a look at how the epoxide group of oxidosqualene is formed. Trends Pharm. Sci. 2005, 26, 335; J. Phys Chem B., 2012, 116, 13857.
Exercise 14.24:
a) The figure below outlines the first, cyclizing phase of the reaction that converts oxidosqualene to lanosterol. However, the diagram is missing electron movement arrows, and intermediates 1-4 are all missing formal charges - fill these in.
First phase (ring formation):
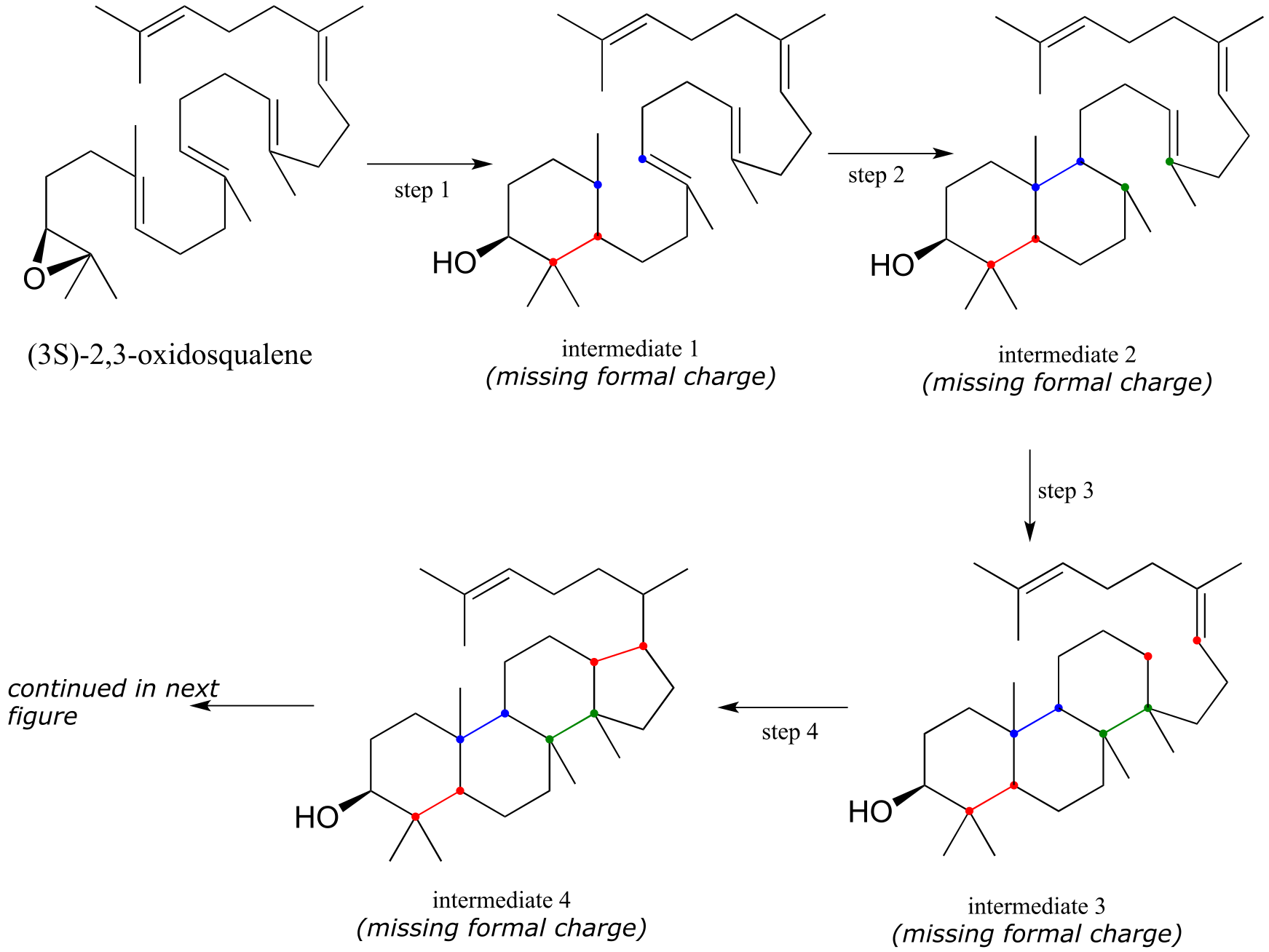
(fig 20)
fig 45
b) Next comes the ‘shifting’ phase of the reaction. Once again, supply the missing mechanistic arrows.
Second phase: rearrangement and deprotonation
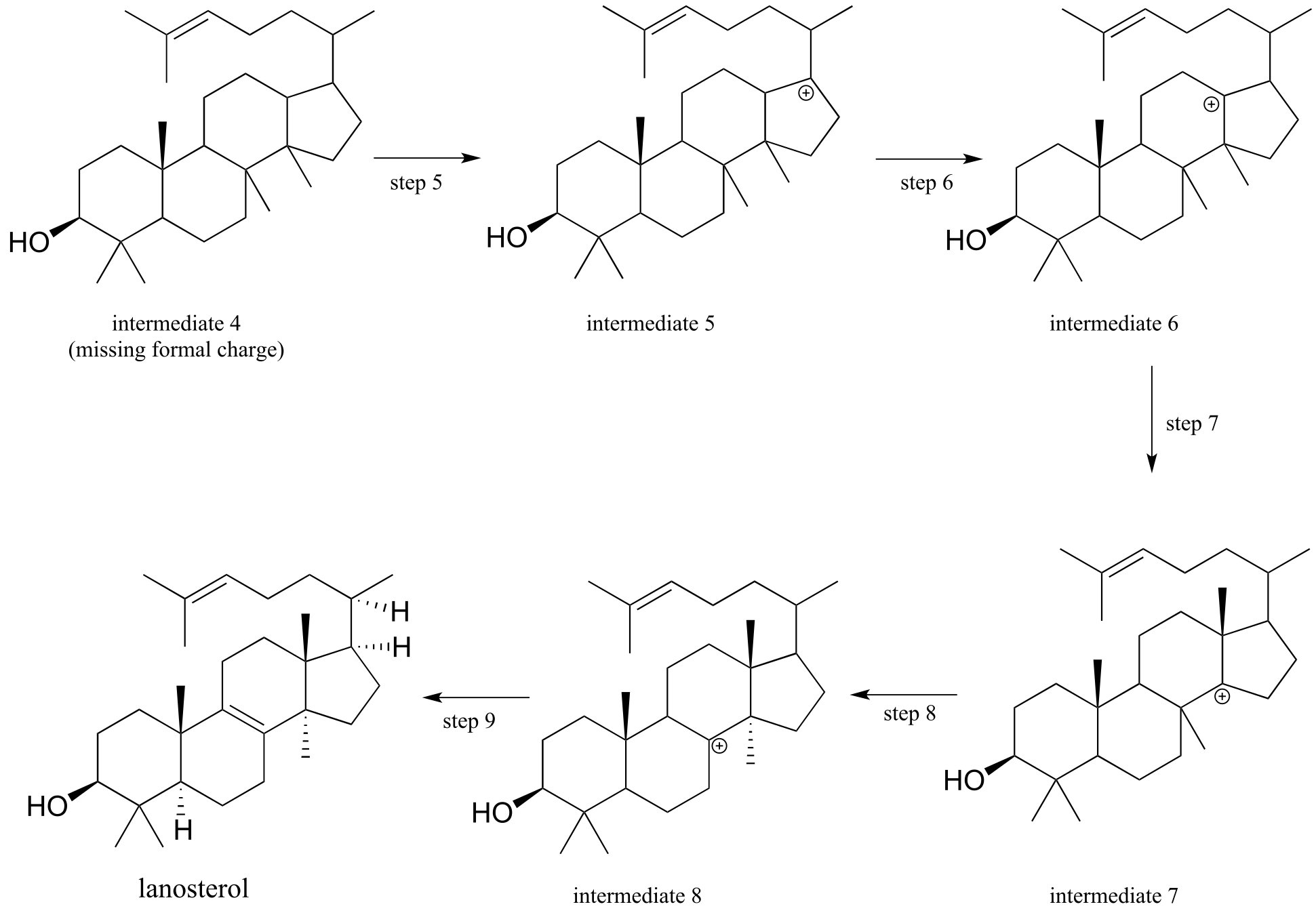
fig 46
c) Look at the first and last steps of the entire process: overall, would you describe this as an electrophilic addition or substitution?
The oxidosqualene cyclization reaction and others like it are truly remarkable examples of the exquisite control exerted by enzymes over the course of a chemical reaction. Consider: an open-chain starting molecule is converted, by a single enzyme, into a complex multiple fused-ring structure with seven chiral centers. Oxidosqualene could potentially cyclize in many different ways, resulting in a great variety of different products. In order for the enzyme to catalyze the formation of a single product with the correct connectivity and stereochemistry, the enzyme must be able to maintain precise control of the conformation of the starting compound and all reactive intermediates in the active site, while also excluding water molecules which could attack at any of the positively charged carbons.
Key learning objectives for this chapter#
Understand why the π bond in a carbon-carbon double bond is more reactive than the σ bond.
Addition
Be able to draw a mechanism for the electrophilic addition of a haloacid to an alkene.
Stereochemistry: understand why nonenzymatic electrophilic addition of a haloacid to an alkene occurs with racemization (both inversion and retention of configuration) at both alkene carbons. Be able to distinguish syn vs anti addition.
Regiochemistry: Be able to predict the regiochemical outcome of an electrophilic addition, based on the relative stability of the two possible carbocation intermediates. Be able to predict when anti-Markovnikov addition is likely to occur.
Be able to predict the product of nonenzymatic addition of water/alcohol to an alkene, including regio- and stereo-chemistry when applicable. Be able to draw complete mechanisms.
Be able to predict the products of nonenzymatic addition of water/alcohol to a conjugated diene or triene, including regio- and stereochemistry when applicable. Be able to draw complete mechanisms, including multiple resonance forms for carbocation intermediates.
Be able to apply your understanding of nonenzymatic alkene addition reactions to draw mechanisms for enzymatic addition reactions. In particular, you should be able to draw mechanisms for biochemical electrophilic addition reactions in which a new carbon-carbon bond is formed.
Elimination
Be able to draw a mechanism for an E1 elimination reaction.
Be able to predict possible E1 reaction products from a common starting compound , taking into account both regiochemistry (Zaitsev’s rule) and stereochemistry.
Be able to recognize and draw a mechanism for biochemical E1 reactions in which
a) the second step is a deprotonation event
b) the second step is a decarboxylation event
Be able to distinguish whether a biochemical elimination reaction is likely to proceed through a E1cb or E1 mechanism, based on the structure of the starting compound.
Isomerization/substitution
Be able to recognize and draw mechanisms for a biochemical electrophilic isomerization reaction (shifting the location of the carbon-carbon double bond).
Be able to recognize and draw mechanisms for a biochemical electrophilic substitution reaction.
Be able to recognize and draw mechanisms for a biochemical electrophilic aromatic substitution reaction, and be able to explain the ring-activating effect (how the carbocation intermediate is stabilized by resonance, usually with lone-pair electrons on either an oxygen or a nitrogen atom).
Be able to recognize when a hydride or alkyl shift is likely to occur with a carbocation reaction intermediate.
Be able to draw a mechanism for a reaction that includes a carbocation rearrangement.
Problems#
P14.1: Draw the major product(s) (including all stereoisomers) that would be expected to result from the nonenzymatic electrophilic addition reactions below. Your product(s) should result from the most stable possible carbocation intermediate. Hint: consider the possibility of thermodynamically favorable rearrangement steps.
a)

b)
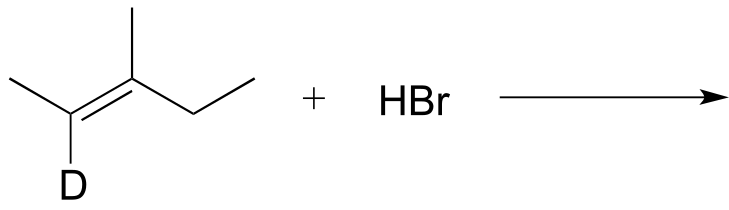
c)
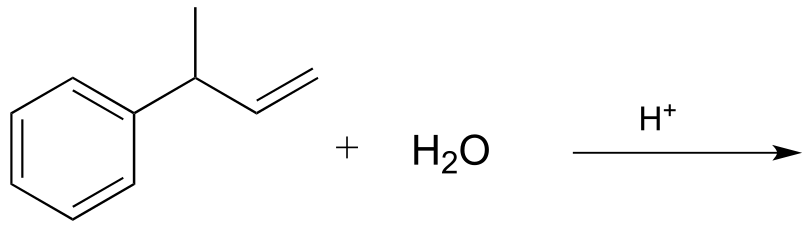
d)
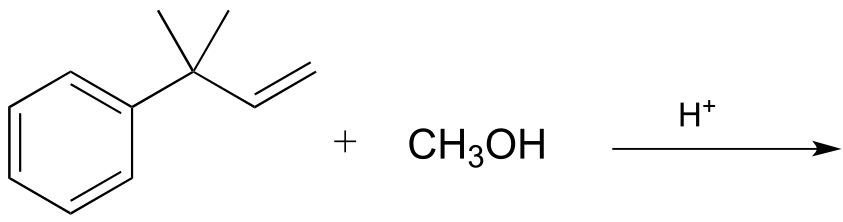
e)
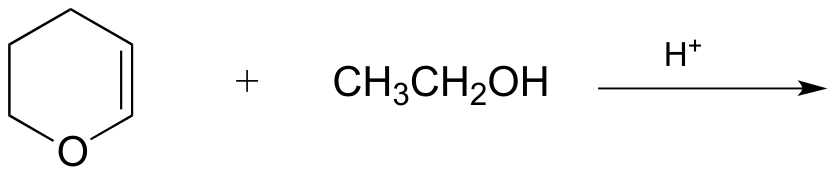
P14.2: Draw likely mechanisms for the nonenzymatic reactions below. Products shown are not necessarily the most abundant for the reaction.
a)
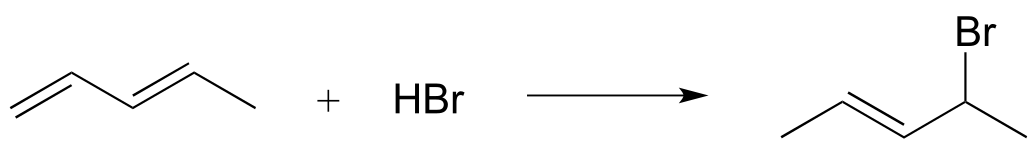
b)
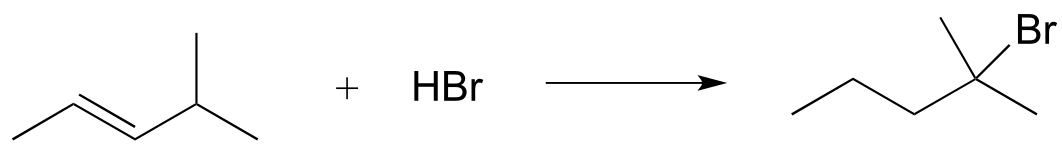
c)
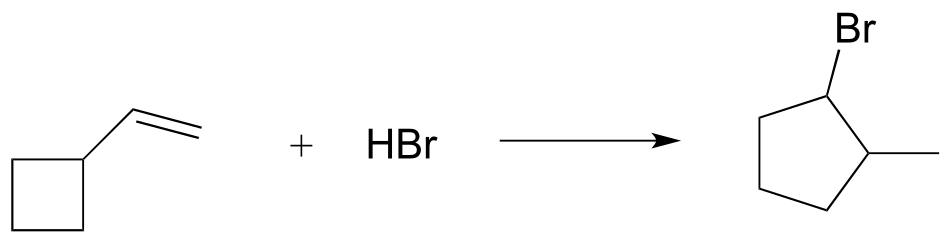
d)
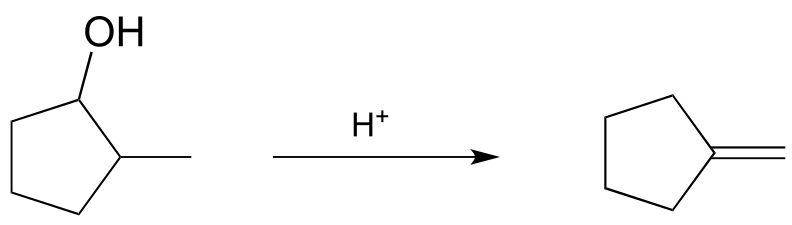
P14.3: Provide mechanisms for the following reactions, both of which are part of an alkaloid synthesis pathway in fungi. (Microbiol. 2005, 151, 2199)
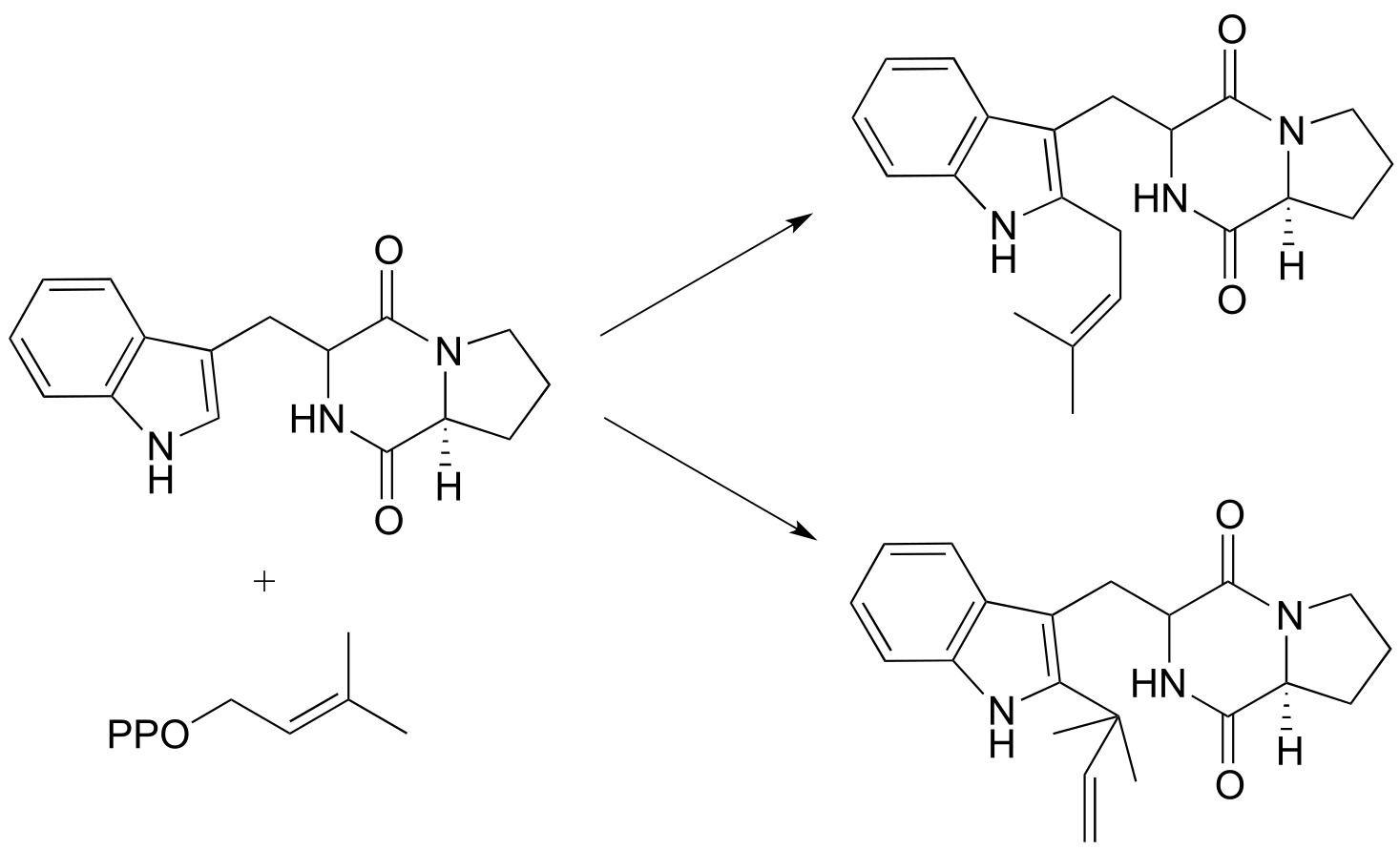
P14.4: Draw a likely mechanism for the reaction below. The product is myrcene, a compound produced by fir trees as a defense against insects. (J. Biol. Chem 1997, 272, 21784)
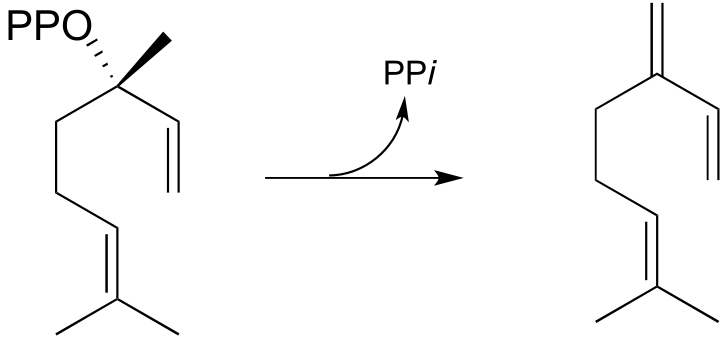
P14.5: Provide a mechanism for the following reaction from the vitamin B12 biosynthetic pathway, and identify the missing participants indicated by questions marks in the figure.
P14.6: A diene molecule synthesized in the laboratory was found to irreversibly inhibit the action of isopentenyl diphosphate isomerase (section 14.3) when the carbon indicated with a dot becomes covalently bonded to a cysteine residue in the enzyme’s active site. Propose a mechanism showing how this could happen. ( J. Am. Chem. Soc. 2005, 127, 17433)
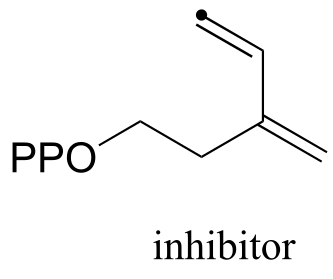
P14.7: Nonenzymatic electrophilic addition of water to alkynes results in the formation of a ketone or an aldehyde, depending on the starting alkyne. A vinylic carbocation is a key intermediate, and the reaction is accelerated with the use of a catalytic amount of strong acid. Predict the product the addition of water to propyne, and draw a mechanism for the reaction.
P14.8: The reaction below is part the pathway by which some bacteria -including the species which cause tuberculosis and leprosy - form distinctive branched-chain fatty acids for incorporation into their cell walls. This enzyme is of interest to scientists as possible targets for new antibiotic drugs. Propose a likely mechanism, and identify the missing participants denoted by questions marks. (J. Biol. Chem. 2006, 281, 4434)
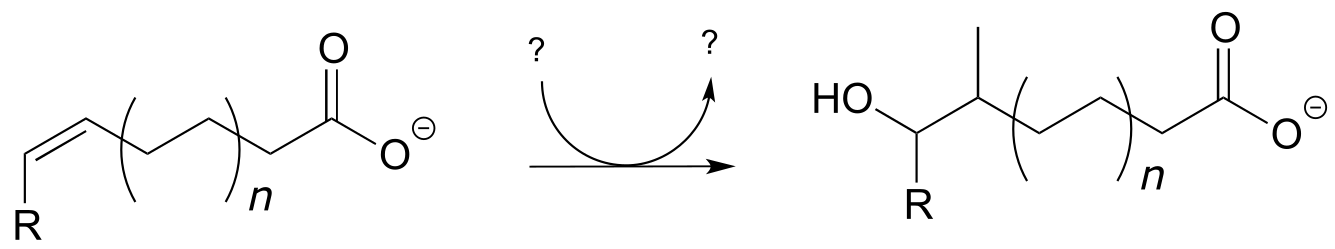
P14.9: Suggest a likely mechanism for this reaction, which is a key step in the synthesis of bacterial cell walls. Your mechanism should show an electrophilic addition, followed by an E1 elimination.
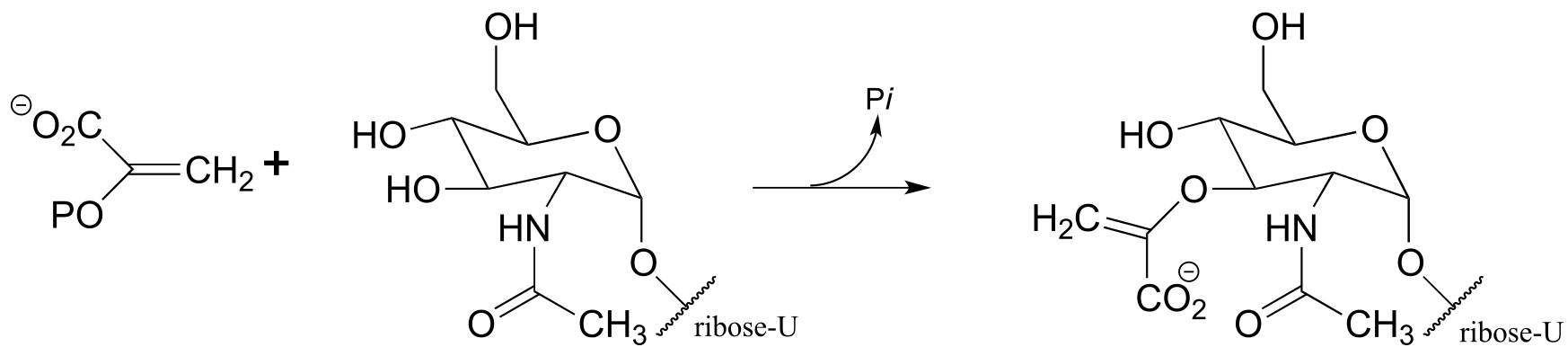
P14.10: Suggest a mechanism for the following reaction, which is part of the pathway by which many microbes synthesize methanopterin, a derivative of the vitamin folic acid. Hint: the mechanism can be described as an electrophilic aromatic substitution with a final decarboxylation step in place of the usual deprotonation step. (J. Biol. Chem. 2004, 279, 39389.
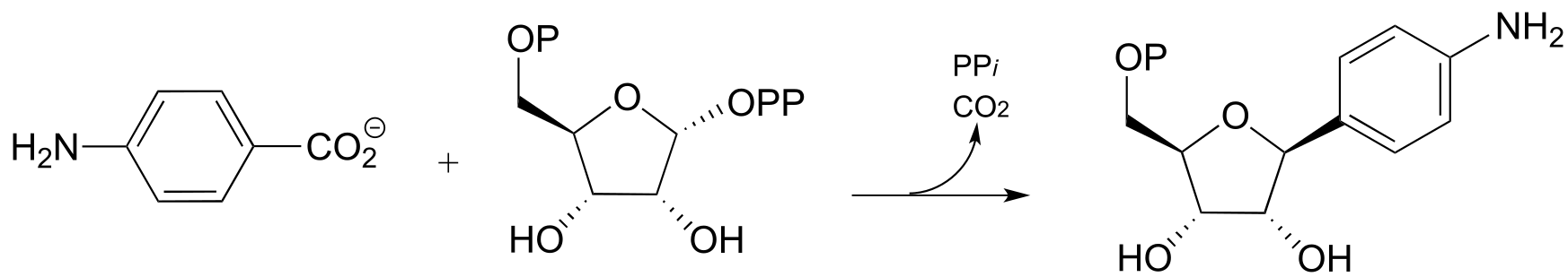
(
P14.11: Researchers investigated the mechanism of the enzyme 3-deoxy-D-manno-octulosonate-8-phosphate synthase by running the reaction with one of the substrates labeled with the 18O isotope (colored red in the scheme below). Consider the two hypothetical results shown below, each pointing to a different mechanism. Both mechanisms involve a carbocation intermediate. (Biochem. Biophys. Res. Commun. 1988, 157, 816)

a) Propose a mechanism that is consistent with result A, in which the 18O label ends up in the ketone group of the organic product.
b) Propose a mechanism that is consistent with result B, in which the 18O label ends up in the inorganic phosphate by-product.
P14.12: Consider the following isomerization reaction (J. Biol. Chem. 1989, 264, 2075):
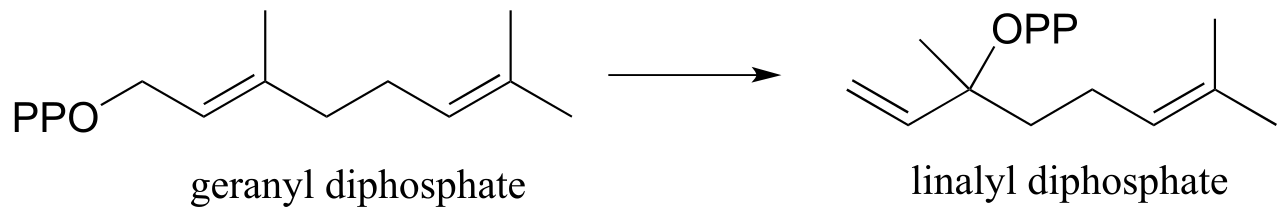
(JBC264, 2075)
a) Suggest a likely mechanism involving a carbocation intermediate.
b) Suggest an isotopic labeling experiment (using substrate labeled with 18O) that could confirm or rule out a alternative, concerted isomerization mechanism (ie. one without formation of a carbocation intermediate). Explain your reasoning.
c) Propose a mechanism for the following reaction (notice that the starting compound is linalyl diphosphate from part (a), drawn in a different conformation). (Arch Biochem Biophys 2003, 417, 203)
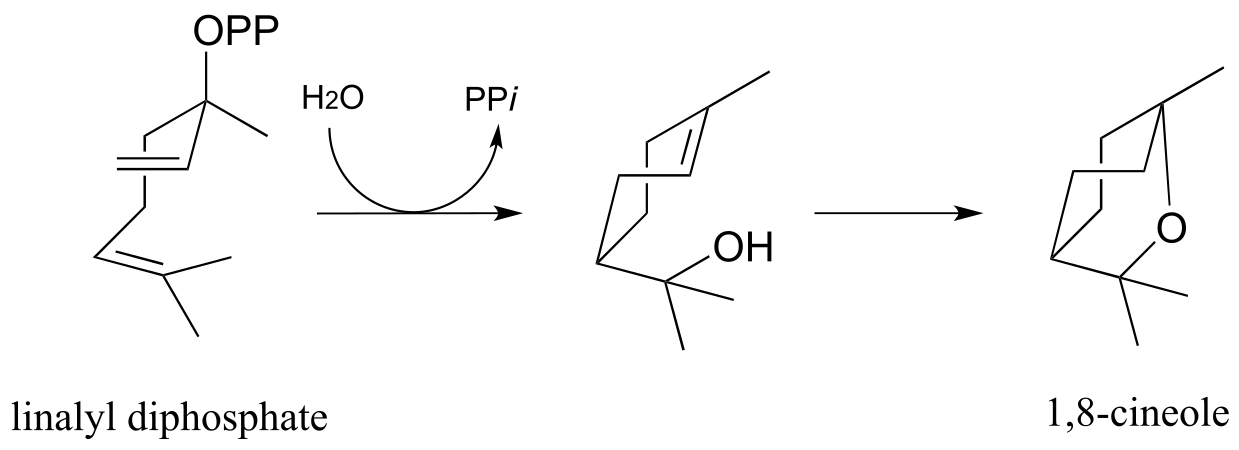
(
For parts d-f, refer to the figure below:
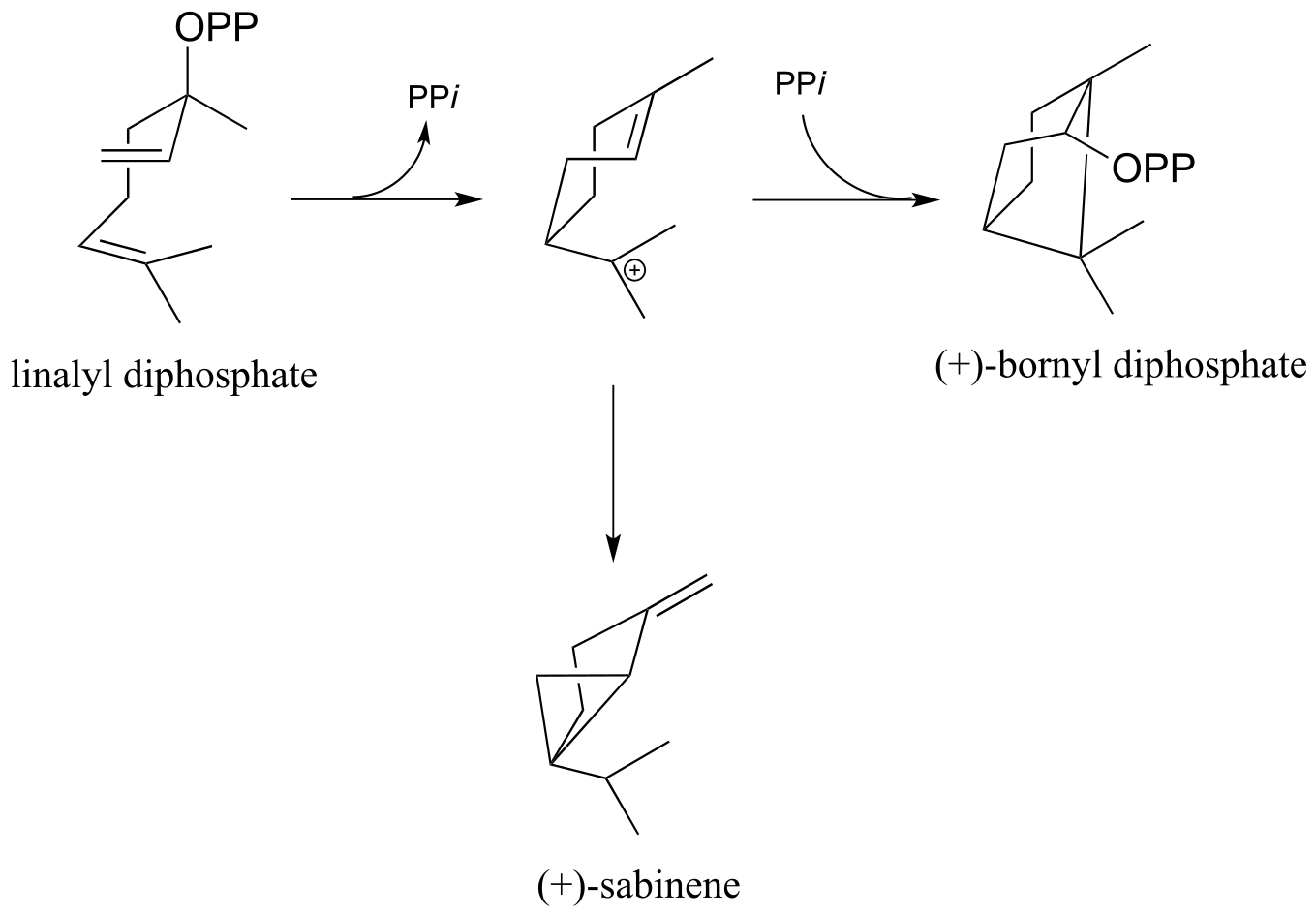
d) Provide mechanisms for the conversion of linalyl diphosphate to (+)-bornyl diphosphate
e) Provide mechanisms for the conversion of linalyl diphosphate to (+)-sabinene.
f) Is the second step in the (+)-bornyl diphosphate pathway (addition of phosphate) a Markovnikov or anti-Markovnikov addition? Explain the regiochemistry of this step in terms of carbocation stability.
P14.13: The two compounds shown below were each treated with HBr, and the products isolated and analyzed by 1H NMR. Use the NMR data provided to determine the structure of both products, then explain the observed regiochemistry of the addition reaction.
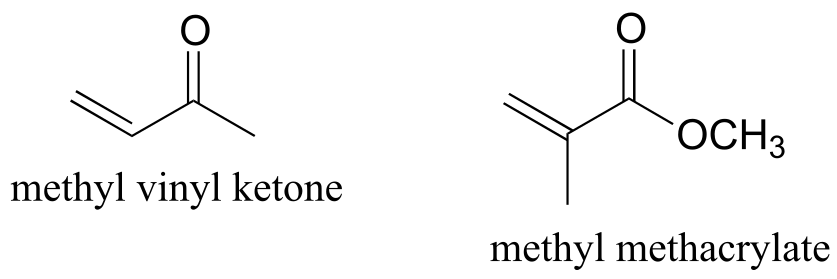
1H-NMR data for product of HBr addition to methyl vinyl ketone:
δ integration splitting
2.2 1.5 s
3.0 1 t
3.5 1 t
1H-NMR data for product of HBr addition to methyl methacrylate:
δ integration splitting
1.3 3 d
2.3 1 sextet
3.5 2 d
3.7 3 s
P14.14: Ketones and aldehydes with a hydroxy group in the position are known to undergo an isomerization reaction known as an acyloin rearrangement:
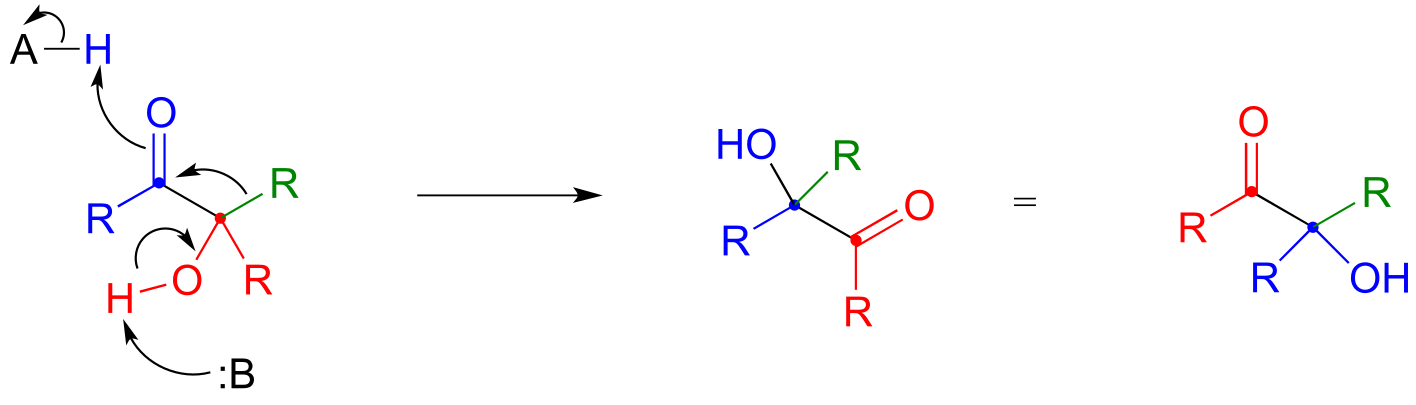
Notice in the general acyloin rearragnement mechanism below, the green alkyl group is shifting from the carbon (red) to the carbonyl carbon (blue). Notice also that this shift does not involve a carbocation intermediate, although a resonance contributor can be drawn in which the carbonyl carbon has a positive charge.
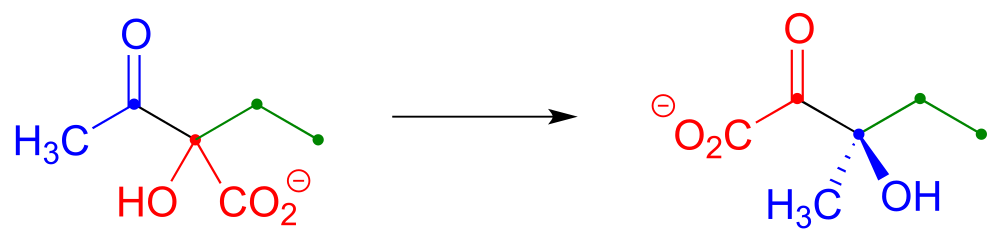
a) Draw a mechanism for this acyloin rearrangement step in the biosynthetic pathway for the amino acid leucine:
b) Draw a mechanism for this acyloin rearrangement step in the isoprenoid biosynthetic pathway in bacteria:

**
**
P14.15: Propose mechanisms for these reactions in the vitamin B12 biosynthetic pathway:
a)
b)
c)
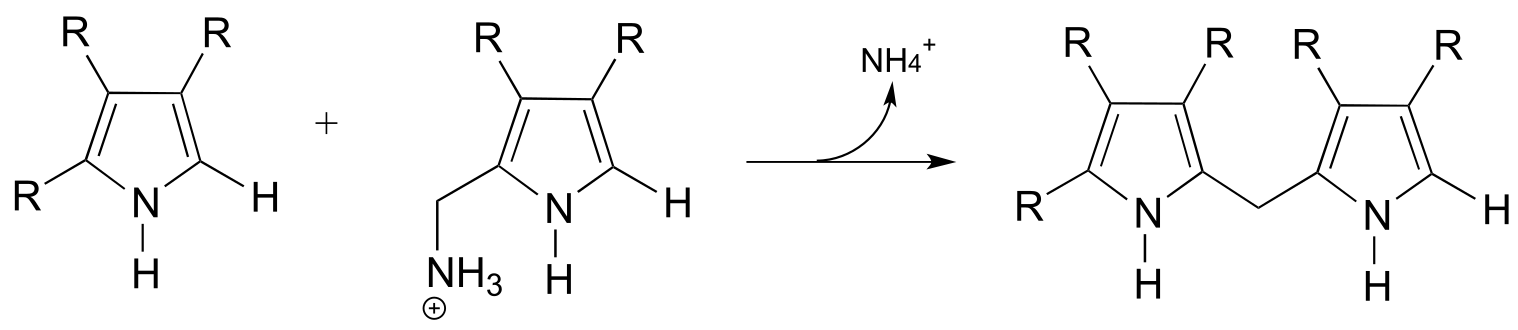
)
P14.16: An early reaction in the biosynthesis of tryptophan can be described as an intramolecular electrophilic aromatic substitution/decarboxylation hybrid, followed by an E1 dehydration (EC 4.1.1.48).
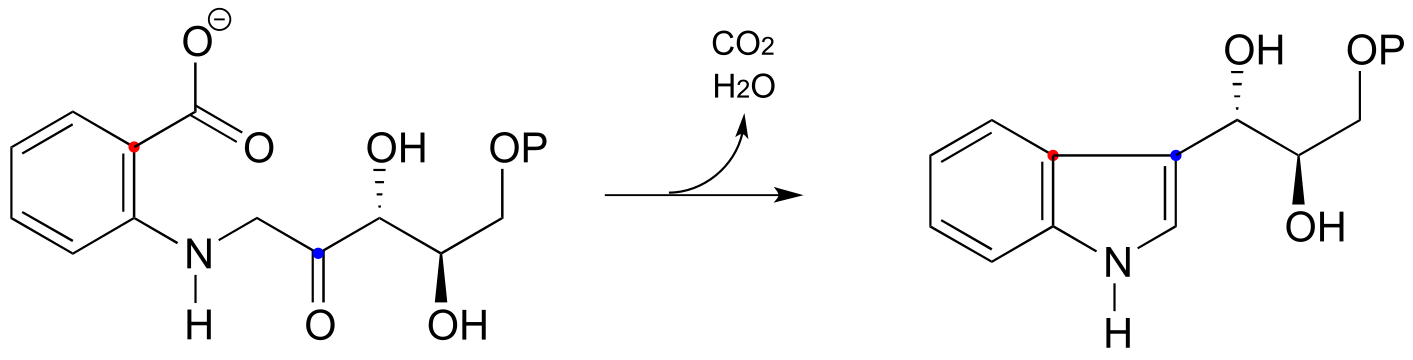
a) Draw a mechanism that corresponds to the verbal description given above. Use resonance structures to show how the nitrogen atom helps to stabilize the carbocation intermediate. Hint: the electrophilic carbon in this case is a ketone rather than a carbocation.
b) What aspect of this reaction do you think helps to compensate for the energetic disadvantage of not having a powerful carbocation electrophile?
c) Again thinking in terms of energetics, what is the ‘driving force’ for the dehydration step?
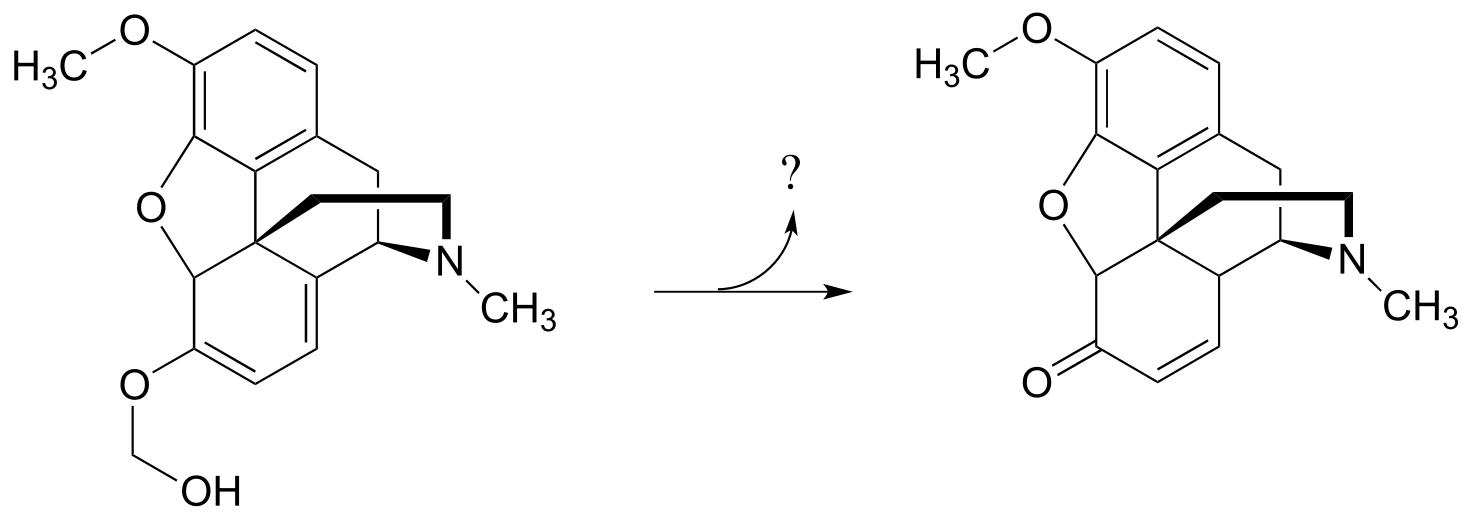
P14.17: Propose a likely carbocation-intermediate mechanism for the following reaction in the biosynthesis of morphine, being sure to identify the structure of the organic compound released in the reaction.(
P14.18: Propose mechanisms for these three electrophilic cyclization reactions. Carbocation rearrangement steps are involved.
a) epi-arisolochene
b) vetispiradiene (Science 1997, 277, 1815)
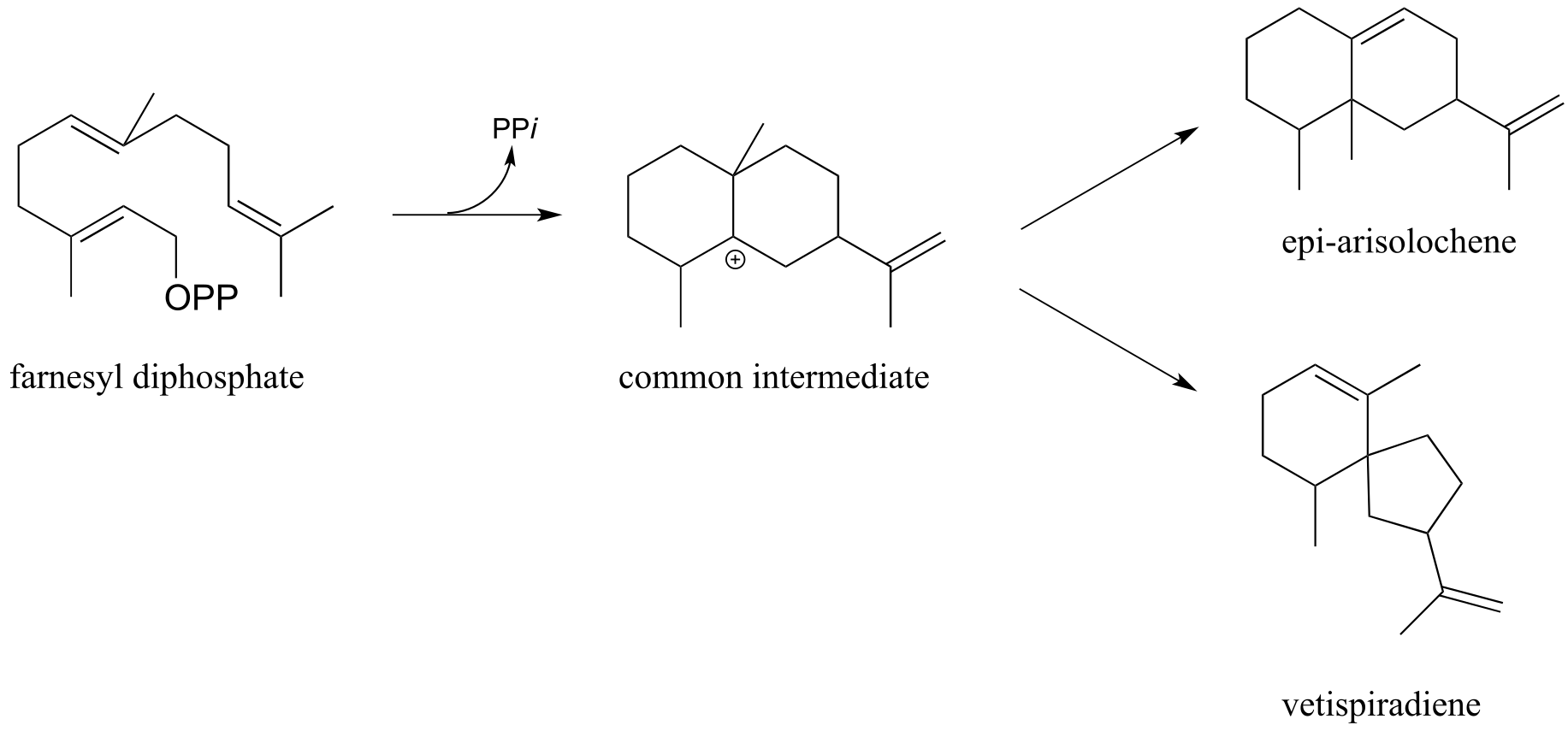
c) pentalenene (Science 1997, 277, 1820)
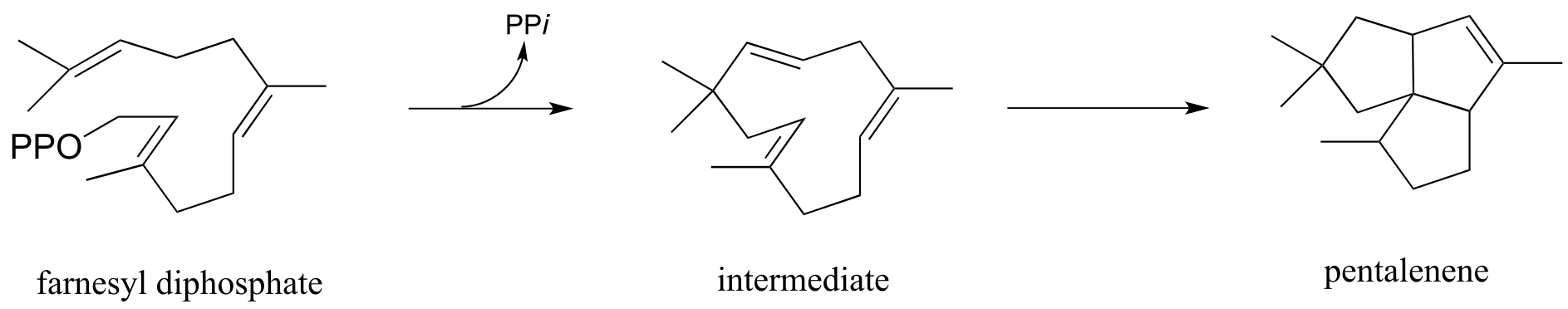
P14.19: Strictosinide, an intermediate in the biosynthesis of the deadly poison strychnine, is formed from two steps: a) intermolecular imine formation, and b) an intramolecular, ring-forming electrophilic aromatic substitution with the imine carbon from step (a) as the electrophile.
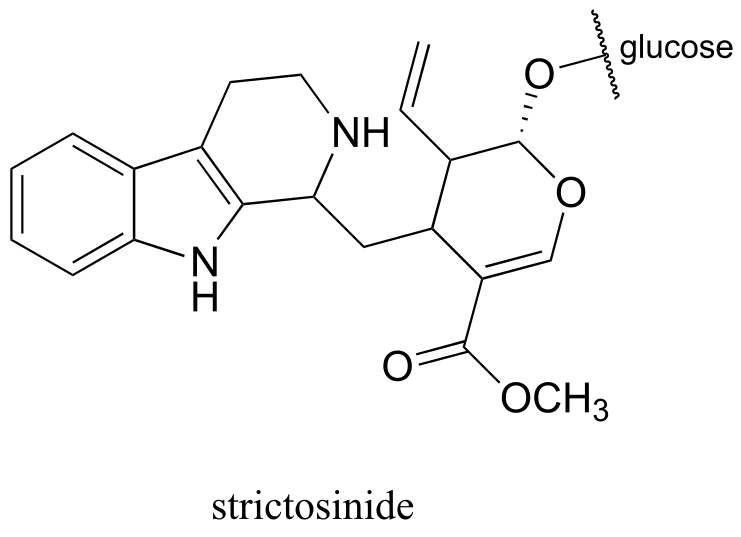
Given this information, predict the two precursors to strictosinide, and draw a mechanism for the reaction described. Hint: use the ‘retro’ skills you developed in chapters 12 and 13.
P14.20: In the introduction to chapter 8, we learned about reactions in which the cytosine and adenine bases in DNA are methylated. In the course of that chapter, we learned how adenine N-methylation occurs in bacteria, but we were not yet equipped to understand cytosine C-methylation, which was the more relevant reaction in terms of human health and development. Now we are: propose a reasonable mechanism for the C-methylation of cytosine.
P14.21: The reaction below has been proposed to proceed via a cyclization step followed by an E1/decarboxylation step (in other words, an E1 mechanism where decarboxylation occurs instead of deprotonation). Draw a mechanism that fits this description, and show the most stable resonance contributors of the two key cationic intermediates.
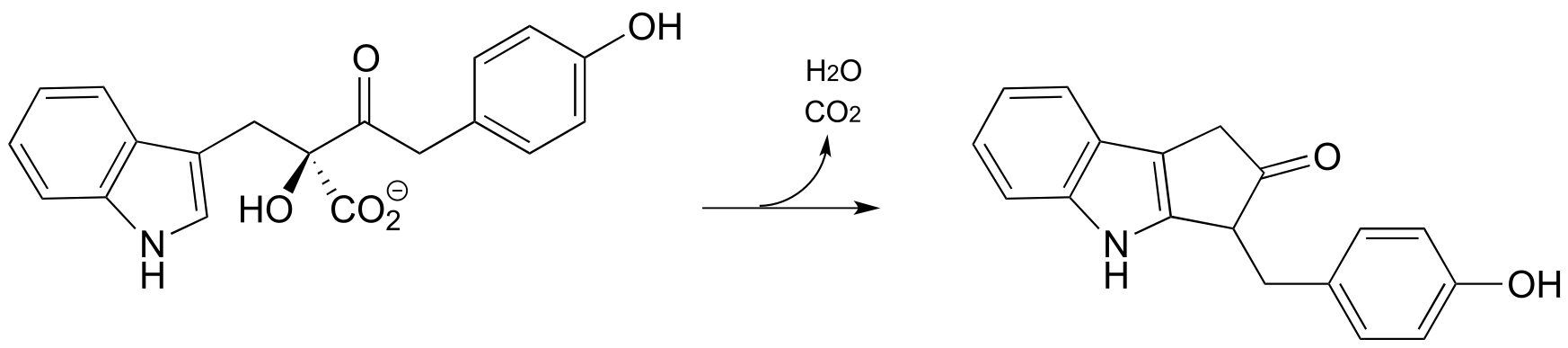
P14.22: The conversion of chorismate to phenylpyruvate is a key transformation in the biosynthesis of phenylalanine. The first step is a concerted electrophilic rearrangement to form prephenate (this step involves a six-membered transition state). Deduce the structure of prephenate, and provide a complete mechanism for the transformation.
(
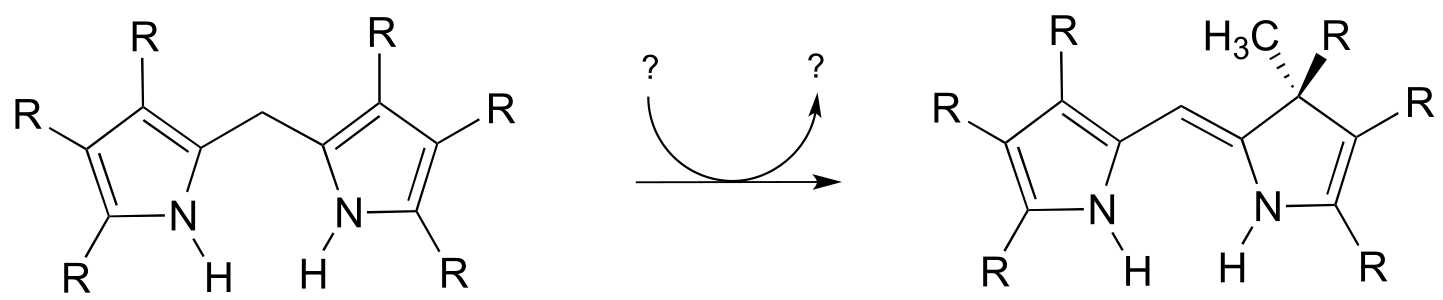
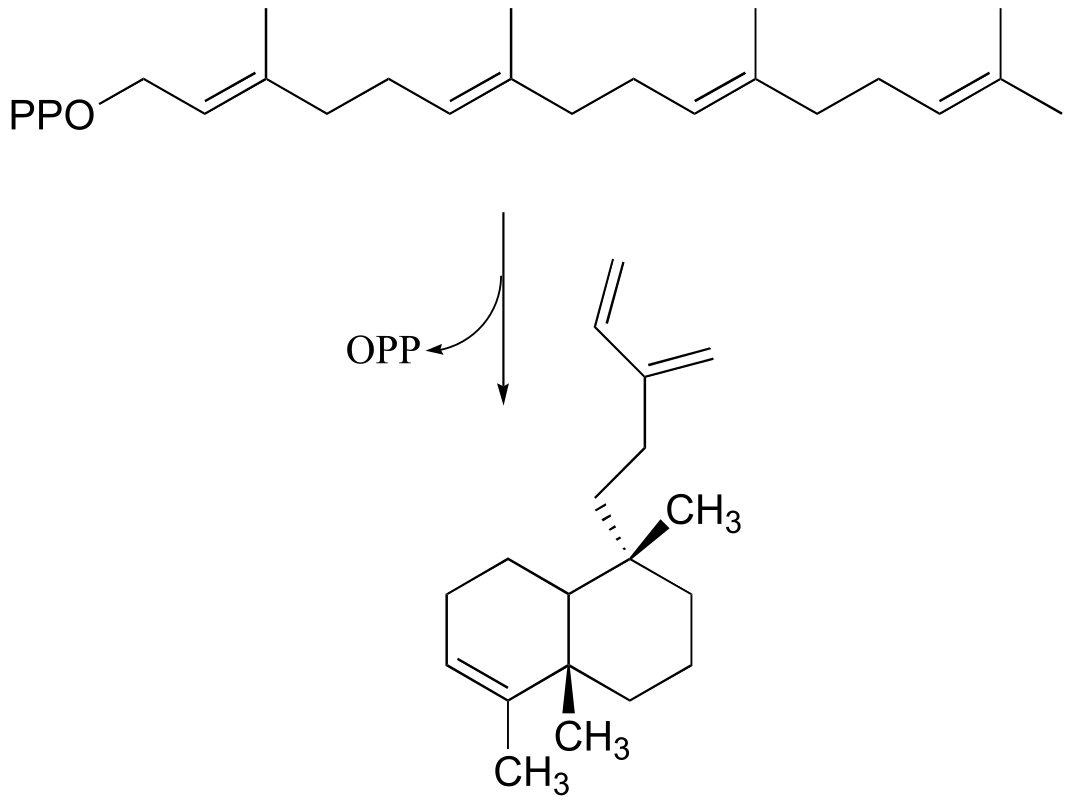
P14.23: Suggest likely a mechanism for the following reaction:
(J. Org. Chem. 2003, 68, 5433)